SIMD-parallel BLAST X-drop alignment implementation. (work in progress...)
- BLAST X-drop DP algorithm (semi-global extension aligmnent of affine-gap penalty); expected to be sensitive enough for most usecases
- Vectorized with SSE4.1 (run on Core2 (2007) or later; 16-bit signed int is used for each cell)
- Linear-to-graph alignment (first developed for vg map subcommand; POA-like method)
- Header-only and C++-compatible
/**
* Aligning ACACTTCTAGACTTTACCACTA to the following graph
* 6
* A
* 0 1 3 5 /7\9
* ACAC-TTGT-AGAC---TTCTA-C-CACTA
* \ / \ / \ /
* ATCC T G
* 2 4 8
*/
/* init score matrix and memory arena */
int8_t const M = 2, X = -3, GI = 5, GE = 1; /* match, mismatch, gap open, and gap extend; g(k) = GI + k + GE for k-length gap */
int8_t const xdrop_threshold = 70;
int8_t const score_matrix[16] = {
/* ref-side */
/* A C G T */
/* A */ M, X, X, X,
/* query- C */ X, M, X, X,
/* side G */ X, X, M, X,
/* T */ X, X, X, M
};
struct dz_s *dz = dz_init(
score_matrix,
GI, GE,
xdrop_threshold
);
/* pack query */
char const *query = "ACACTTCTAGACTTTACCACTA";
struct dz_query_s const *q = dz_pack_query_forward(
dz,
query, /* char const *seq: query sequence */
strlen(query) /* length */
);
/* init node array */
struct dz_forefront_s const *ff[10] = { 0 };
/* fill root: node 0 */
ff[0] = dz_extend(
dz, q,
dz_root(dz), 1, /* struct dz_forefront_s const *ff[], uint64_t n_ffs; incoming forefront and degree; dz_root(dz) for root node */
"ACAC", strlen("ACAC"), 0 /* reference-side sequence, its length, and node id */
);
/* branching paths: 1, 2 -> 3 */
ff[1] = dz_extend(dz, q, &ff[0], 1, "TTGT", strlen("TTGT"), 1);
ff[2] = dz_extend(dz, q, &ff[0], 1, "ATCC", strlen("ATCC"), 2);
ff[3] = dz_extend(dz, q, &ff[1], 2, "AGAC", strlen("AGAC"), 3); /* "&ff[1], 2" indicates ff[1] and ff[2] are incoming nodes */
/* insertion: -, 4 -> 5 */
ff[4] = dz_extend(dz, q, &ff[3], 1, "T", strlen("T"), 4);
ff[5] = dz_extend(dz, q, &ff[3], 2, "TTCTA", strlen("TTCTA"), 5); /* "&ff[3], 2" indicates ff[3] and ff[4] are incoming nodes */
/* SNVs: 6, 7, 8 -> 9 */
ff[6] = dz_extend(dz, q, &ff[5], 1, "A", strlen("A"), 6);
ff[7] = dz_extend(dz, q, &ff[5], 1, "C", strlen("C"), 7);
ff[8] = dz_extend(dz, q, &ff[5], 1, "G", strlen("G"), 8);
ff[9] = dz_extend(dz, q, &ff[6], 3, "CACTA", strlen("CACTA"), 9);
/* detect max */
struct dz_forefront_s const *max = NULL;
for(size_t i = 0; i < 10; i++) {
if(max == NULL || ff[i]->max > max->max) { max = ff[i]; }
}
/* traceback */
struct dz_alignment_s const *aln = dz_trace(
dz,
ff[9]
);
printf("ref_length(%u), query_length(%u), score(%d), path(%s)\n", aln->ref_length, aln->query_length, aln->score, aln->path);
for(size_t i = 0; i < aln->span_length; i++) {
struct dz_path_span_s const *s = &aln->span[i];
printf("node_id(%u), subpath_length(%u), subpath(%.*s)\n",
s->id,
s[1].offset - s[0].offset,
s[1].offset - s[0].offset, &aln->path[s->offset]
);
}
/* clean up */
dz_destroy(dz);Running the example above:
% make && ./example
gcc -O3 -march=native -o example example.c
ref_length(23), query_length(22), path(======X=======D========)
node_id(0), subpath_length(4), subpath(====)
node_id(1), subpath_length(4), subpath(==X=)
node_id(3), subpath_length(4), subpath(====)
node_id(5), subpath_length(5), subpath(==D==)
node_id(7), subpath_length(1), subpath(=)
node_id(9), subpath_length(5), subpath(=====)
%
- Only supports nucleotide sequences -> tentative support added.
- Newer instruction support is not planned (e.g. AVX2 and AVX-512; keep it as simple as possible).
- Node length to be shorter than 32768 / M.
Query sequence is first packed into an array of SIMD vectors with its encoding converted from ASCII to 2-bit. It is required to enable fast unaligned load for each subsequence and to ensure the vectorized load does not touch outside of the array. It does not construct precalculated score profile vectors, which is common for SIMD-parallel alignment algorithms such as Rognes, Farrar, and Myers' bit-parallel. Rather the score profile vector is calculated in an on-the-fly manner by 16-element shuffle instruction in the DP loop. The on-the-fly calculation is executed in a single CPU cycle and expected to be slightly faster than loading a precalculated vector from memory or cache.
For the detailed description of the BLAST X-drop DP algorithm, see the document by @elucify. The following a simplified illustration of POA-like graph-capable extension of the algorithm.
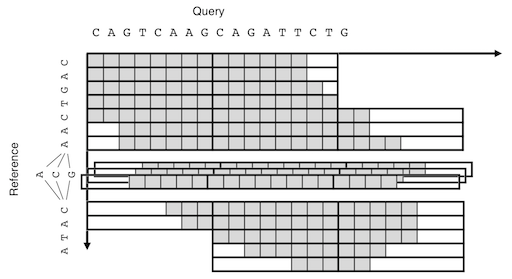
An illustration of the SIMD-parallel X-drop DP aligning a sequence to a graph.
The fill-in function takes an array of preceding nodes. The function first merges the incoming vectors by taking the maximum for each DP cell. The DP matrix is then extended row by row until the end of the reference sequence. A reference sequence with negative length is treated as its reverse-complemented scaned from the last character, for example, (seq, len) = (&"AACG"[4], -4) gives the same result as ("CGTT", 4).
The X-drop condition is tested on each vector rather than on each cell. A vector is dropped only when all the cell have their values below the current maximum minus X. This conservative scheme ensures the algorithm to evaluate all the cells that are evaluated in the original non-vectorized X-drop DP. Thus it is expected the sensitivity will not be degraded by this modification.
The maximum score position is updated when newer cell value takes greater than or equal to the current maximum. It result in reporting longer path for the reference direction. On the other hand, the shortest path is reported for the query direction. (The former cmpeq behavior can be overwritten by defining dz_cmp_max macro).
The maximum-scoring node is not kept tracked internally; it must be tracked by caller. The reason the library is unaware of maximum-scoring node is that the scores cannot be compared between two diverging paths (i.e. there is no way to know what the maximum of another leaf node is from a leaf when each node-filling function does not have knowledge about the whole tree structure).
The traceback function takes a node (forefront object) pointer. It first determines the maximum-scoring cell for the node then begin traceback from the cell. It automatically roll the node chain up to the root and returns a pair of alignment path and mapping section array.
Other encoding such as (A, C, G, T) = (1, 2, 3, 4) can be used for input by modifying the dz_pack_query_{forward, reverse} functions, which is in particular done by re-filling the conversion table in the function. Currently the 16-element table is composed of 0, A, 0, C, T, U, 0, G, 0, 0, 0, 0, 0, 0, qN, 0, which is overlap of former and latter half columns of the ASCII table: 0, A, 0, C, 0, 0, 0, G, 0, 0, 0, 0, 0, 0, N, 0 and 0, 0, 0, 0, T, U, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0 (fortunately each position does not conflict). A similar table is used for the reference side, too.
The query-sequence packing part can be removed when the 2-bit encoding (A, C, G, T) = (00, 01, 10, 11) is used for the query sequence and the region is allocated with a margin longer than the SIMD vector length. It will slightly improve the performance but complicates the input sequence preparation.
The on-the-fly score profile calculation algorithm is unable to be applied to larger score matrices such as BLOSUM and PAM. These large matrices, which cannot be kept on SIMD registers, are better handled by the precalculated score profile vector algorithm. The modified alogirthm would be much similar to the Rognes's composition. The array should be allocated in the local memory arena just after the struct dz_query_s object by dz_mem_malloc so that the overhead of allocation and access to be as small as possible. For typical protein score matrix we need to build 20 lanes, each of which length is equal to the query length rounded up by the SIMD register width.
We can also extend the precalculated score matrix algorithm to k-mer x k-mer matrix (e.g. 4096 x 4096 for k = 6), though, the number of vectors, 4096, is too huge to be filled on initialization. The vectors are also unable to be kept inside the last-level cache (4096 x 1 kbp = 4 MB > 3 MB). I believe we would get better performance by not creating the precalculated vectors but just fetching each score from the matrix when it is needed (in the unparallelized manner). It consumes a lot of CPU cycles waiting for the data to arrive however the situation is inevitable when we adopt such huge score matrix. So it would be even reasonable to hand the cache over to the DP matrix. It is also worth considering to keep locality of the score matrix with regards to the overlapping k-mers by adopting de Bruijn sequence ordering for each axis but the expected cache hit rate will still be low (I have not verified it yet).
In contrast, the 4 x k-mer matrix to align Nanopore signal to consensus sequences is treated in almost the same way as the original 4 x 4 score matrix. The difference is that we still need the precalculated score profile vectors because the entire score matrix cannot be kept on the SIMD register. However, the vector construction is not a heavy task because the number of lanes is small.
I would like to give thank to Erik Garrison for his description on the internals of the vg toolkit, and insightful comments about alignment algorithms. I also thank Toshiaki Katayama and DBCLS staffs for holding 3rd RDF summit at Kyoto where this work has begun.
MIT
Copyright 2018, Hajime Suzuki