JBrowse_Configuration_Guide
This page provides a comprehensive reference guide for configuring JBrowse. If you find something that is missing or inaccurate, please feel very free to edit it!
Note: this is a reference guide. It is not meant to be read from beginning to end. If this is your first time setting up JBrowse, you probably want to read the Quick-start tutorial first, and then consult this guide when you need information on specific things you want to do with your JBrowse.
Check out the new JBrowse FAQ page for more tips on setup and configuration.
Also see the JBrowse Desktop guide here.
At the most basic level, setting up JBrowse consists of:
- Placing a copy of the JBrowse directory somewhere in the web-servable part of your server's file system (often
/var/wwwby default) - Running the JBrowse setup script to install a few server-side dependencies
- Running one or more server-side scripts to create a directory containing a JBrowse-formatted copy of your data.
Both the JBrowse code and these data files must be in a location where the web server can serve them to users. Then, a user pointing their web browser at the appropriate URL for the index.html file in the JBrowse directory will see the JBrowse interface, including sequence and feature tracks reflecting the data source.
Reference sequence data should be added first (using prepare-refseqs.pl`), followed by annotation data. Once all of annotation data has been added, use generate-names.pl to make the feature names searchable.
- Install build prerequisites, plus make and a C compiler. On Ubuntu, you could do this with:
sudo apt-get install zlib1g-dev libpng-dev libgd2-noxpm-dev build-essential
Some other things that sometimes need to be manually installed if your setup.sh is failing includes these
sudo apt-get install libexpat-dev libxml2-dev libdb-dev
If you need a web server you can add apache2 to the list
-
Download JBrowse onto your web server.
-
Unpack JBrowse into a directory that is served by your web browser. On many systems, this defaults to
/var/wwwor/var/www/htmlfor apache2
cd /var/www/html
unzip JBrowse-*.zip
Make sure you have permissions to write to the contents of the jbrowse/ directory you have just created.
-
Run the automated-setup script,
./setup.sh, which will attempt to install all of JBrowse's (modest) prerequisites for you in thejbrowse/directory itself. Note thatsetup.shshould not be run as root or withsudo. -
Visit http://your.machine.address/jbrowse/index.html?data=sample_data/json/volvox. If you can see the included Volvox example data, you are ready to configure JBrowse to show your own data! The Getting Started with JBrowse Tutorial provides a very basic step-by-step guide to formatting your own data, and in-depth configuration reference information can be found on this page.
Note: if there is an error installing the perl pre-requisites, and you get an error in your setup.log such as /home.local/username/.cpanm/build.log: No such file or directory at /loader/0x10b108f0/App/cpanminus/script.pm line 224.
Then you can clear out your users locallib with rm -rf ~/.cpanm and re-run setup.sh. Only do this if you are not concerned about your personal cpanm folder in the first place. Otherwise, you can use your system's cpanm to install jbrowse pre-requisites with cpanm . inside the jbrowse directory.
To upgrade an existing JBrowse (1.3.0 or later) to the latest version, simply move its data directory (and jbrowse.conf or jbrowse_conf.json if you are using it) into the directory of a newer JBrowse (and with v1.13.0 or later also run setup.sh after each modification of jbrowse.conf or jbrowse_conf.json in case you use any plugins), and the new JBrowse will display that data.
To upgrade a 1.2.x JBrowse, copy its data directory into the new JBrowse directory, and point your browser at compat_121.html in the new JBrowse directory, instead of index.html. Or, if desired, you could simply overwrite index.html with compat_121.html.
If you are upgrading from a version of JBrowse older than 1.2.0, a fresh installation is required.
JBrowse supports two configuration formats, a JSON-based format and a GBrowse-like textual format that is easier to edit and maintain by hand than JSON. Sites can use either format, or a mixture of both. The default shipped configuration of JBrowse uses both: jbrowse.conf in the main JBrowse directory for global settings, and trackList.json in each data directory for dataset-specific configuration in JSON, and tracks.conf in the data directory for dataset-specific configuration in .conf format.
The JSON configuration format was the first format supported by JBrowse, and is easy for software programs to read and modify. Before version 1.11.0, this was the only format supported by JBrowse.
As an example, the trackList.json file might have something like this. Here is an example of a BAM track
{
"tracks": [
{
"urlTemplate" : "volvox-sorted.bam",
"storeClass" : "JBrowse/Store/SeqFeature/BAM",
"type" : "JBrowse/View/Track/Alignments2",
"label" : "BAM_track",
"key" : "My BAM track"
"style": { "color": "red" }
}
]
}
The specifics of this config are not essential, we are specifying an array of tracks in a trackList.json style, and each track is an object that includes some parameters like the urlTemplate to refer to the location of the BAM file on the server relative to the data directory, the color of the features, etc.
- Nested objects are specified using typical JSON format, using curly brackets
- Booleans and numbers should remain unquoted
- Functions do remain quoted however e.g. "style": { "color": "function() { /* your code here */ }" }
- JSON strings should not contain line breaks (see Text .conf format for info on multiline callbacks)
- Configuration values can be stored in both jbrowse_conf.json or in trackList.json (or conf files) i.e. the trackList.json does not only have to contain tracks, can contain other config entries
JBrowse 1.11.0 introduced support for a new text-based configuration format that users of GBrowse will find very familiar, since its design borrows heavily from GBrowse’s configuration syntax. It is fairly comfortable to hand-edit, but rather inconvenient for automated tools to work with. To get the best of both worlds, JBrowse supports both formats.
This text configuration format can be used to specify
- general configuration options (i.e. jbrowse.conf 1)
- track-specific options (i.e. tracks.conf 2)
- standalone files with extra code (i.e. functions.conf 3).
The text format has several benefits, including the ability to specify multi-line callbacks. Example:
# BAM track with a new callback
[tracks.mytrack]
storeClass = JBrowse/Store/SeqFeature/BAM
type = JBrowse/View/Track/Alignments2
urlTemplate = myfile.bam
key = My BAM track
style.color = function(feature) {
/* comment */
return 'red';
}
- Comments should start with #
- The section labels, e.g. [tracks.testtrack] defines an identifier for the track named testtrack, so you should not have dots in your identifier, e.g. don't use something like this [tracks.test.track]
- Don't quote the values in the file, e.g key=My BAM track, not key="My BAM track"
- Nested values can specified using 'dot' notation, e.g. "style.color"
- A "section" can be specified with square brackets, e.g. [trackMetadata] will create the config variable trackMetadata and the values in the section are added to it.
- Extra JSON values can be specified in the conf file using the syntax json:{...} (see 4 for example)
- Very large .conf files (thousands of lines) files can take longer to parse than equivalent JSON
- An array of values can be built up over multiple lines. NOTE: A quirk of the format is that there cannot be more than 4 spaces before the + sign in each item. Example:
[trackMetadata]
sources =
+ data/mymeta.csv
+ data/more_meta.csv
- Comments inside callbacks can use the /* */ format but not the // format
- All lines of a multi-line callback should be spaced away from the left-most column, including the closing bracket (see the style.color example above)
- There should be no blank lines inside a multi-line callback
- Refer to 5 for more info on multi-line functions
When your web browser loads a page containing JBrowse, and JBrowse starts, the following steps are done
- In index.html, read the URL params (e.g. query params like &data= and &tracks, &loc=, etc.)
- In index.html, create a JSON blob using URL params and pass them to the Browser.js constructor which you can see on index.html
- In Browser.js, the constructor is a JSON blob that becomes the "root configuration object"
- In Browser.js, mix the root config with the _defaultConfig object
- In _defaultConfig, the default is to include both jbrowse_conf.json and jbrowse.conf config files
- In jbrowse.conf, the default is to include {dataRoot}/trackList.json {dataRoot}/tracks.conf
This is how you eventually get the trackList.json and tracks.conf files from your data directory loaded. Note that the &data=blah URL parameter becomes the dataRoot config parameter, so dataRoot would be "blah" in that case.
The configuration system then merges all this information, e.g. from the URL params, from the Browser constructors, the defaultConfigs, the jbrowse.conf, the jbrowse_conf.json, the trackList.json, the tracks.conf, and any files that the trackList.json or tracks.conf files themselves include, into a single config.
Generally this happens all seamlessly, and both the text-based .conf format and .json config files can co-exist. That is because anything that can be written as a .conf file can also be written as a .json, they are both parsed on the client side into config objects.
The reference sequences on which the browser will display annotations, and which provide a common coordinate system for all tracks. At a close enough zoom level, the sequence bases are visible in the "Reference Sequence" track.
The exact interpretation of "reference sequence" will depend on how you are using JBrowse. For a model organism genome database, each reference sequence would typically represent a chromosome or a contig. Before any feature or image tracks can be displayed in JBrowse, the reference sequences must be defined using the prepare-refseqs.pl formatting tool.
JBrowse displays a dropdown selector for changing reference sequences.
For JBrowse versions 1.6.3 to 1.8.1, if more than 30 reference sequences are loaded, this selector is not shown by default. To force JBrowse to show the reference sequence dropdown selector, set refSeqDropdown: true in the configuration. This can be done in any configuration file, e.g. index.html, jbrowse_conf.json, or data/trackList.json.
In JBrowse version 1.9.0 and later, the reference sequence dropdown menu is always displayed. However, if there are too many reference sequences to practically display in the dropdown selector, the first portion of the sequence list is shown in the dropdown selector, along with a message stating that there are more reference sequences than can be shown. The maximum number of reference sequences in the selector is set by the refSeqSelectorMaxSize configuration variable, and defaults to 30.
The ordering of reference sequences in the selector is configurable using the refSeqOrder configuration variable.
Supported values for refSeqOrder include
namename descendinglengthlength descending-
false/null/0to disable any sorting -
by_listto manually specify a list of reference sequences in the selector
One instance in which refSeqOrder is particularly useful is in displaying annotations on early-stage, incomplete genomic assemblies: to display the N biggest contigs in the assembly in the reference sequence selector dropdown, one can set refSeqOrder to 'length descending', and set refSeqSelectorMaxSize to N.
If you set refSeqOrder to by_list, you can then set refSeqOrderList to set the exact order of the reference sequence list.
Example (in data/tracks.conf)
[GENERAL]
refSeqOrder = by_list
refSeqOrderList =
+ ctgB
+ ctgA
+ ctgAprime
+ bethsCrazyBananasContig
+ ctgAZed
This script is used to format sequence data for use by JBrowse, and must be run before adding other tracks. In addition to formatting the sequence data, this script creates a track called "DNA" that displays the reference sequence. The simplest way to use it is with the --fasta option, which uses a single sequence or set of reference sequences from a FASTA file:
bin/prepare-refseqs.pl --fasta <fasta file> [options]
If the file has multiple sequences (e.g. multiple chromosomes), each sequence will become a reference sequence by default. You may switch between these sequences by selecting the sequence of interest via the pull-down menu to the right of the large "zoom in" button.
You may use any alphabet you wish for your sequences (i.e., you are not restricted to the nucleotides A, T, C, and G; any alphanumeric character, as well as several other characters, may be used). Hence, it is possible to browse RNA and protein in addition to DNA. However, some characters should be avoided, because they will cause the sequence to "split" - part of the sequence will be cut off and and continue on the next line. These characters are the hyphen and question mark. Unfortunately, this prevents the use of hyphens to represent gaps in a reference sequence.
In addition to reading from a fasta file, prepare-refseqs.pl can read sequences from a gff file or a database. In order to read fasta sequences from a database, a config file must be used.
Syntax used to import sequences from gff files:
bin/prepare-refseqs.pl --gff <gff file with sequence information> [options]
Syntax used to import sequences with a config file:
bin/prepare-refseqs.pl --conf <config file that references a database with sequence information> --[refs|refid] <reference sequences> [options]
Syntax used to import a indexed fasta(i.e. a fasta file where you run `samtools faidx yourfile.fa` which outputs yourfile.fa.fai)
bin/prepare-refseqs.pl --indexed_fasta yourfile.fa
This will copy yourfile.fa and yourfile.fa.fai to the data directory
| Option | Value |
|---|---|
| fasta, indexed_fasta, twobit, gff, sizes, or conf | Path to the file that JBrowse will use to import sequences. With the fasta and gff options, the sequence information is imported directly from the specified file. With the sizes option, a tab delimited file with chromosome names and lengths is used, but no sequence information is added. With the conf option, the specified config file includes the details necessary to access a database that contains the sequence information. Exactly one of these three options must be used. With indexed_fasta, the samtools faidx yourfile.fa must be run before hand. With twobit, the twobit file will automatically be copied into your data directory. |
| out | A path to the output directory (default is 'data' in the current directory) |
| seqdir | The directory where the reference sequences are stored (default: <output directory>/seq) |
| noseq | Causes no reference sequence track to be created. This is useful for reducing disk usage. |
| refs | A comma-delimited list of the names of sequences to be imported as reference sequences. This option (or refid) is required when using the conf option. It is not required when the fasta or gff options are used, but it can be useful with these options, since it can be used to select which sequences JBrowse will import. |
| refids | A comma-delimited list of the database identifiers of sequences to be imported as reference sequences. This option is useful when working with a Chado database that contains data from multiple different species, and those species have at least one chromosome with the same name (e.g. chrX). In this case, the desired chromosome cannot be uniquely identified by name, so it is instead identified by ID. This ID can be found in the 'feature_id' column of 'feature' table in a Chado database. |
Note: the prepare-refseqs.pl --sizes chrom.sizes option is maybe underappreciated. You can technically run jbrowse without any sequence data loaded, simply a set of chromosomes and their sizes. The chrom.sizes file simply can contain two column tab seperated list of chromosome names and their lengths.
JBrowse has several different tools that can be used to convert range-based annotation data (genes, transcripts, etc) to range-indexed sets of static JSON files that are very fast for JBrowse to access. Each of these tools also adds a track configuration stanza to the trackList.json configuration file in its output directory.
- flatfile-to-json.pl - import GFF3 and BED files (recommended for new users)
- biodb-to-json.pl - import from a Bio::DB::SeqFeature::Store database (recommended for users with existing databases)
- ucsc-to-json.pl - import UCSC database dumps (.sql and .txt.gz)
Each run of this script formats a single track for JBrowse. A flat file is a data set that exists entirely in a single file. For this script, the file must be a GFF3, BED, or GenBank text file.
Basic usage:
bin/flatfile-to-json.pl --[gff|gbk|bed] <flat file> --tracklabel <track name> [options]
For a full list of the options supported by flatfile-to-json.pl, run it with the --help option
bin/flatfile-to-json.pl --help
Example
flatfile-to-json.pl \
( --gff <GFF3 file> | --bed <BED file> | --gbk <GenBank file> ) \
--trackLabel <track identifier> \
[ --trackType <JS Class> ] \
[ --out <output directory> ] \
[ --key <human-readable track name> ] \
[ --className <CSS class name for displaying features> ] \
[ --urltemplate "http://example.com/idlookup?id={id}" ] \
[ --arrowheadClass <CSS class> ] \
[ --noSubfeatures ] \
[ --subfeatureClasses '{ JSON-format subfeature class map }' ] \
[ --clientConfig '{ JSON-format style configuration for this track }' ] \
[ --config '{ JSON-format extra configuration for this track }' ] \
[ --thinType <BAM -thin_type> ] \
[ --thicktype <BAM -thick_type>] \
[ --type <feature types to process> ] \
[ --nclChunk <chunk size for generated NCLs> ] \
[ --compress ] \
[ --sortMem <memory in bytes to use for sorting> ] \
[ --maxLookback <maximum number of features to buffer in gff3 files> ] \
[ --nameAttributes "name,alias,id" ] \
The --trackLabel parameter is the only required parameter, and is the "id" to refer to your track by. The displayed name is also whatever --trackLabel is unless --key is specified, in which case, whatever --key is will be used as the displayed name.
By default the output is in a folder called data in your current working directory, or whatever is specified by --out
Using --trackType CanvasFeatures is generally useful since CanvasFeatures are newer than the default HTMLFeatures (aka FeatureTrack)
This script uses a config file to produce a set of feature tracks in JBrowse. It can be used to obtain information from any database with appropriate schema, or from flat files. Because it can produce several feature tracks in a single execution, it is useful for large-scale feature data entry into JBrowse.
Basic usage:
bin/biodb-to-json.pl --conf <config file> [options]
For a full list of the options supported by biodb-to-json.pl, run it with the --help option, like:
bin/biodb-to-json.pl --help
This script uses data from a local dump of the UCSC genome annotation MySQL database. To reach this data, go to hgdownload.cse.ucsc.edu and click the link for the genome of interest. Next, click the "Annotation Database" link. The data relevant to ucsc-to-json.pl (*.sql and *.txt.gz files) can be downloaded from either this page or the FTP server described on this page.
Together, a *.sql and *.txt.gz pair of files (such as cytoBandIdeo.txt.gz and cytoBandIdeo.sql) constitute a database table. Ucsc-to-json.pl uses the *.sql file to get the column labels, and it uses the *.txt.gz file to get the data for each row of the table. For the example pair of files above, the name of the database table is "cytoBandIdeo". This will become the name of the JBrowse track that is produced from the data in the table.
In addition to all of the feature-containing tables that you want to use as JBrowse tracks, you will also need to download the trackDb.sql and trackDb.txt.gz files for the organism of interest.
Basic usage:
bin/ucsc-to-json.pl --in <directory with files from UCSC> --track <database table name> [options]
Hint: If you're using this approach, it might be convenient to also download the sequence(s) from UCSC. These are usually available from the "Data set by chromosome" link for the particular genome or from the FTP server.
For a full list of the options supported by ucsc-to-json.pl, run it with the --help option, like:
bin/ucsc-to-json.pl --help
Feature tracks can be used to visualize localized annotations on a sequence, such as gene models, transcript alignments, SNPs and so forth.
JBrowse HTMLFeatures tracks, the default legacy track type for range-based features, have many available options for customization, not all of which are available from the command-line formatting scripts. Below is a comprehensive list of configuration options for HTMLFeatures tracks. HTMLFeatures tracks are also referred to as trackType: "FeatureTrack" or "type": "FeatureTrack".
| Option | Value |
|---|---|
yScalePosition |
Position of the y-axis scale indicator when the track is zoomed far enough out that density histograms are displayed. Can be "left", "right", or "center". Defaults to "center". |
maxFeatureScreenDensity |
Maximum density of features to display on the screen. If this is exceeded, will display either feature density histograms (if available), or a "too many features to show" message. The units of this number are features per screen width in pixels. Defaults to 0.5. |
description |
Comma-separated list of fields in which to look for the description of a feature. Case-insensitive. If set to false or null, no feature description will be shown. Defaults to 'note, description'. |
maxDescriptionLength |
Maximum length, in characters, for displayed feature descriptions. |
minSubfeatureWidth |
Minimum width, in pixels, of the top-level feature for JBrowse to attempt to display its subfeatures. Default 6. |
menuTemplate |
Optional menu configuration for right-click menus on features. Can be as large and complicated as you want. See Customizing_Right-click_Context_Menus below. If set to null or false, disables feature right-click menus. |
hooks→create |
JavaScript function that creates the parent feature HTML element and returns it. By default this is: function(track,feature) { return document.createElement('div'); }, which creates an HTML div element. |
hooks→modify |
JavaScript function that can be used to modify the feature HTML element in any way desired. If set, the function is called with the track object, feature object, and feature HTML element as arguments (signature: function(track, feature, featDiv)). |
style→featureScale |
Minimum zoom scale (pixels/basepair) for displaying individual features in the track. Not set by default, and overrides the maxFeatureScreenDensity. |
style→className |
CSS class for parent features. Defaults to "feature". |
style→subfeatureClasses |
Key-value pairs of CSS classes for subfeatures, organized by feature type. Example: { "CDS" : "transcript-CDS","UTR" : "transcript-UTR" } |
style→featureCss |
Text string of additional CSS rules to add to features. Example: "border-color: purple; background-color: yellow;" |
style→arrowheadClass |
CSS class of the strand arrowheads to show for this feature. Defaults to 'arrowhead'. If set to null, no arrowhead will be drawn. |
style→histScale |
Scale (pixels per basepair) below which the track will attempt to draw feature density histograms instead of features, if available. By default, this is set to 4 times the average feature density (features per basepair) of the track. |
style→label |
Comma-separated list of case-insensitive feature tags to use for showing the feature's label. The first one found will be used. Default 'name,id'. |
style→labelScale |
Scale (pixels per basepair) above which feature labels (names) will be shown. By default, this is set to 30 times the average feature density (features per basepair) of the track. |
style→descriptionScale |
Scale (pixels per basepair) above which long feature descriptions will be shown. By default, this is set to 170 times the average feature density (features per basepair) of the track. |
style→description |
Comma-separated list of case-insensitive feature tags to check for the feature's long description. The first one found will be used. Default 'note,description'. If blank no description is used. |
style→showLabels |
If set to true, feature labels may be shown. Defaults to true. Set this to false to disable display of feature labels. |
maxHeight |
Maximum height, in pixels, that the track is allowed to grow to. When it reaches this height, features that stack higher than this will not be shown, and a "Max height reached" message will be displayed. Default 600 pixels. |
showNoteInAttributes |
If set to true, show the feature's "Note" attribute as a regular attribute in the feature detail dialog. This is mostly useful for projects that want the blue description text on a feature to be different from the feature's Notes attribute, but still display the Notes attribute in the detail dialog |
topLevelFeatures |
Specifies which feature types should be considered "top-level" for this track. For example, if you have a track with gene->mRNA->CDS features, but for some reason want to only display the mRNA features, you can set topLevelFeatures=mRNA. Can also be an array of string types, or a function callback that returns an array of types. Default: all features are displayed. Added in 1.14.0 |
Introduced in JBrowse 1.10.0, the new JBrowse CanvasFeatures tracks are faster and easier to configure than HTMLFeatures tracks.
| Option | Description |
|---|---|
maxHeight |
Maximum height, in pixels, that the track is allowed to grow to. When it reaches this height, features that stack higher than this will not be shown, and a "Max height reached" message will be displayed. Default 600 pixels. |
style→showLabels |
If true, allows feature labels to be shown if features are not too dense on the screen. Default true. |
style→showTooltips |
If true, allows feature name tooltips to be shown. Default true. |
displayMode |
'normal', 'compact', or 'collapsed'. Sets the initial display mode of the track. Default 'normal'. |
style→featureScale |
Minimum zoom scale (pixels/basepair) for displaying individual features in the track. Not set by default, and overrides the maxFeatureScreenDensity. |
maxFeatureScreenDensity |
Maximum density of features to display on the screen. If this is exceeded, will display either feature density histograms (if available), or a "too many features to show" or "too much data to show" message. The units of this number are features per screen width in pixels. Defaults to 0.5. If increased to ~6 then it should pretty much always go away |
glyph |
JS class name of the glyph to use for each feature. By default, it tries to guess for each feature based on its type attribute, and uses JBrowse/View/FeatureGlyph/Box if it cannot find something better. Can be a callback with signature (feature), returning a string class name. |
menuTemplate |
Optional menu configuration for right-click menus on features. Can be as large and complicated as you want. See Right-click_Context_Menus below. If set to null or false, disables feature right-click menus. |
style→maxDescriptionLength |
Maximum length, in characters, of long feature descriptions, for glyphs that support them. Default 70. |
style→color |
Basic color of features. Most glyphs interpret this as the fill color of the rectangle they draw. Color syntax is the same as that used for CSS, specified at http://dev.w3.org/csswg/css-color/. Default 'goldenrod'. |
style→mouseovercolor |
Color of the overlay drawn on top of features when the mouse hovers over them. Color syntax is the same as that used for CSS, specified at http://dev.w3.org/csswg/css-color/. Default rgba(0,0,0,0.3), which is semi-transparent black. |
style→borderColor |
Color of the borders drawn around boxes in glyphs. Color syntax is the same as that used for CSS, specified at http://dev.w3.org/csswg/css-color/. Default varies from glyph to glyph. |
style→borderWidth |
Width of the borders drawn around boxes in glyphs. Default 0.5 if borderColor specified |
style→height |
Height in pixels of glyphs. Default value varies from glyph to glyph. |
style→marginBottom |
Margin space to leave below each feature when arranging them in the view. Default 2 pixels. |
style→strandArrow |
If true, allow glyphs to draw strand arrowheads on features that are stranded. Default true. |
style→label |
Comma-separated list of case-insensitive feature tags to use for showing the feature's label. The first one found will be used. Default 'name,id'. |
style→textFont |
Font used for feature labels. Same format as CSS font rules. Default 'normal 12px Univers,Helvetica,Arial,sans-serif'. |
style→textColor |
Color of feature labels. Color syntax is the same as that used for CSS, specified at http://dev.w3.org/csswg/css-color/. Default 'black'. |
style→text2Color |
Color of feature descriptions. Color syntax is the same as that used for CSS, specified at http://dev.w3.org/csswg/css-color/. Default 'blue'. |
style→text2Font |
Font used for feature descriptions. Same format as CSS font rules. Default 'normal 12px Univers,Helvetica,Arial,sans-serif'. |
style→description |
Comma-separated list of case-insensitive feature tags to check for the feature's long description. The first one found will be used. Default 'note,description'. If blank no description is used. |
style→connectorColor |
Color of the connecting line drawn between boxes in glyphs that draw segments (like the Segments, ProcessedTranscript, and Gene glyphs). Color syntax is the same as that used for CSS, specified at http://dev.w3.org/csswg/css-color/. Default '#333'. |
style→connectorThickness |
Thickness in pixels of the connecting line drawn between boxes in glyphs that draw segments (like the Segments, ProcessedTranscript, and Gene glyphs). Default 1. |
style→utrColor |
Color of UTR regions drawn by ProcessedTranscript and Gene glyphs. Color syntax is the same as that used for CSS, specified at http://dev.w3.org/csswg/css-color/. Defaults to be a color that complements the current style→color value (calculated using a bit of color theory). |
subParts |
Comma-separated list of feature type tags that will be drawn as subparts of parent features. Defaults to all features for Segments glyphs, and 'CDS, UTR, five_prime_UTR, three_prime_UTR' for ProcessedTranscript glyphs. |
transcriptType |
For Gene glyphs, the feature type tag that indicates that a subfeature is a processed transcript. Defaults to 'mRNA'. |
labelTranscripts |
For Gene glyphs, if true, draw a label with the transcript's name beside each transcript, if space permits. Default true. |
style→transcriptLabelColor |
For Gene glyphs, the color of transcript name labels. Color syntax is the same as that used for CSS, specified at http://dev.w3.org/csswg/css-color/. Default 'black'. |
style→transcriptLabelFont |
For Gene glyphs, the font used for transcript name labels. Same format as CSS font rules. Default 'normal 10px Univers,Helvetica,Arial,sans-serif'. |
impliedUTRs |
Introduced in JBrowse 1.10.5. If true, indicates that UTRs are not present in the feature data, but should be inferred from the overlap of exon and CDS features in ProcessedTranscript and Gene glyphs. Default false. Can be a callback. |
maxFeatureGlyphExpansion |
A factor to expand the glyphs by so that if subfeatures go outside the bounds of the parent feature, they will still be rendered. Default: 500bp/current scale. |
inferCdsParts |
If a single CDS span covers the whole gene except the UTRs, then it is drawn as though it only covers the exon parts (not the introns). Default: false. Added in 1.12.3 |
disableCollapsedClick |
Disables the click action when track is collapsed. Default: false. Added in 1.13.0 |
enableCollapsedMouseover |
Enables the mouseover action when track is collapsed. Default: false. Added in 1.13.0. See the ChromHMM track in volvox sample browser for example of mouseover on the collapsed track |
topLevelFeatures |
Specifies which feature types should be considered "top-level" for this track. For example, if you have a track with gene->mRNA->CDS features, but for some reason want to only display the mRNA features, you can set topLevelFeatures=mRNA. Can also be an array of string types, or a function callback that returns an array of types. Default: all features are displayed. Added in 1.14.0 |
ItemRgb |
If set to true, the RGB colors specified in BigBed or BED files will be used for the feature's background color. Default true. Added in 1.14.0 |
Note: the "compact" displayMode for CanvasFeatures tracks uses style->height and multiplies it by 0.35 to create the compact view. Therefore, if you adjust style->height to a smaller default value, then you can create "ultra compact" visualizations.
Unlike HTML-based feature tracks, canvas-based feature tracks don't use modify and create hooks. Instead, the glyph variable, and all of the style variables, support customization callbacks.
All style callbacks are like:
function( featureObject, variableName, glyphObject, trackObject ) { return '#ffcccc'; }
And the glyph callback is like:
function( featureObject ) { return 'JBrowse/View/FeatureGlyph/<glyphclassname>'; }
So, for example, if you wanted to customize a CanvasVariants track to color all homozygous variants blue, and all heterozygous variants red, you could set something like:
[tracks.myvcf]
...
variantIsHeterozygous = function( feature ) {
var genotypes = feature.get('genotypes');
for( var sampleName in genotypes ) {
try {
var gtString = genotypes[sampleName].GT.values[0];
if( ! /^1([\|\/]1)*$/.test( gtString) && ! /^0([\|\/]0)*$/.test( gtString ) )
return true;
} catch(e) {}
}
return false;
}
style.color = function( feature, variableName, glyphObject, track ) {
return track.config.variantIsHeterozygous(feature) ? 'red' : 'blue';
}
Note: the multiline callbacks are only enabled in the tracks.conf form. See Text Configuration Format (.conf) for more considerations about this format.
| Option | Description |
|---|---|
subfeatureDetailLevel |
Set the level of detail of the View details box. If set to 1, only displays one level of subfeatures for example. Default: 0 which displays all subfeature levels. Added in 1.12.3 |
Starting in JBrowse version 1.11.3, the ability to customize parts of the 'View details' Pop-ups was added. This lets you specify functions that have the format fmtDetailValue_* or fmtDetailField_* to either change the value section of an attribute in the config, or the fieldname of an attribute in the config.
Here is an example in tracks.conf format for formatting the "Name" field by adding a link to it:
[tracks.mygff]
key = My Gene Track
storeClass = JBrowse/Store/SeqFeature/NCList
type = FeatureTrack
fmtDetailValue_Name = function(name) {
return "<a href='http://www.example.com?featurename="+name+"'>"+name+"</a>";
}
Note: It is also easy to specify these methods in trackList.json format.
{
"key": "My Gene Track",
"storeClass" : "JBrowse/Store/SeqFeature/NCList",
"type" : "FeatureTrack",
"label": "mygff",
"fmtDetailValue_Name": "function(name) { return '<a href=\"http://www.example.com?featurename='+name+'\">'+name+'</a>'; }"
}
Addendum: If the field has multiple values (e.g. multiple DBXrefs or GO terms), then the callback will receive an array as it's argument, and then you can also return an array which indicates that each element will be formatted inside its own <div>. In this case you will check that the input is an array, because it will also be called on the individual elements too. For example:
"fmtDetailValue_links": "function(links) { if(Array.isArray(links)) { return links; } else return `<a href=\"${link}\">${link}</a>`; }"
This shows that you could, in essence, pre-process the array if you wanted, but the same callback is then called on the individual elements, so you handle both these cases.
In JBrowse 1.11.5, some additional customizations to the pop-up boxes were added.
- The ability to access the feature data was added to the callback signature of the fmtDetailValue_* functions. Example:
fmtDetailValue_Name = function(name, feature) {
/* only add links to the top-level feature */
if(feature.get('type')=='mRNA') {
return name + ' [<a href=http://www.ncbi.nlm.nih.gov/gquery/?term='+name+'>Search NCBI</a>]';
}
}
-
The ability to customize the 'About track' popups was added. These callbacks are named fmtMetaValue_* and fmtMetaField_*
-
The ability to customize mouseover descriptions of the fieldnames was also added. These callbacks are named fmtDetailDescription_* and fmtMetaDescription_*
-
The ability to remove a field from the popup was added. You can do this by returning null from a fmtDetailField_* and fmtMetaField_* callback;
Beginning with JBrowse 1.5.0, the left-clicking behavior of feature tracks (both HTMLFeatures and CanvasFeatures) is highly configurable. To make something happen when left-clicking features on a track, add an onClick option to the feature track's configuration.
In the example configuration below, left-clicks on features will open an embedded popup window showing the results of searching for that feature's name in NCBI's global search, and "search at NCBI" will show in a tooltip when the user hovers over a feature with the mouse:
"tracks": [
{
"label" : "ReadingFrame",
"category" : "Genes",
"class" : "dblhelix",
"key" : "Frame usage",
"onClick" : {
"label": "search at NCBI",
"url": "http://www.ncbi.nlm.nih.gov/gquery/?term={name}"
}
}
...
For details on all the options supported by onClick, see Click Configuration Options.
Note: the style→linkTemplate variable can also be used to specify a URL for left-click on features, but this is a legacy option.
The onClick→label attribute from the previous section is used as the mouse-over description for features on the HTMLFeatures and CanvasFeatures tracks.
In JBrowse 1.11.6, the onClick→label attribute was extended further to allow the mouse-over description to be customized using callbacks and template strings.
Example for CanvasFeatures, allows full HTML tooltips. Here the {name} template is automatically filled in with the feature info:
"onClick": {
"label" : "<div style='font:normal 12px Univers,Helvetica,Arial,sans-serif'>Feature name: {name}</div>",
"title" : "{name} {type}",
"action": "defaultDialog"
}
Example for HTMLFeatures, which only allows plain text descriptions but can support newlines (essentially uses <div title="..."> for mouseover).
"onClick": {
"label": "Feature name {name}\nFeature start {start}\nFeature end {end}",
"title" : "{name} {type}",
"action": "defaultDialog"
}
Example using a callback (for either HMTLFeatures or CanvasFeatures), using this.feature to access the feature details
"onClick": {
"label": "function() { return 'Feature name: '+this.feature.get('name'); }",
"title" : "{name} {type}",
"action": "defaultDialog"
}
Note: on CanvasFeatures, the action "defaultDialog" isn't necessary, but it is necessary for HTMLFeatures to keep the default dialog (as of writing, 1.11.6).
Also note: The "label" which is used in the text for mouseover will be used for the title of any popup by default, so you might also specify a different title.
Also also note: Your mouseover will crash if your features do not have an ID or name, even if you coded the mouseover to not use ID or name.
Canvas-based feature tracks (CanvasFeatures) support an optional histograms configuration subsection that can contain a definition for a second datastore that holds quantitative data (usually either coverage depth or feature density) to be displayed when zoomed further out than featureScale (or if featureScale is not set, the scale determined by the store's feature density divided by maxFeatureScreenDensity). This is often used for BAM coverage on Alignments2 tracks using the histograms.urlTemplate and histograms.storeClass arguments.
Example track
[ tracks.mytrack ]
histograms.storeClass = JBrowse/Store/SeqFeature/BigWig
histograms.urlTemplate = coverage.bw
storeClass = JBrowse/Store/SeqFeature/BAM
urlTemplate = file.bam
type = Alignments2
Feature tracks can be configured to display a context menu of options when a user right-clicks a feature item. Here is an example of a track configured with a multi-level right-click context menu:
{
"feature" : [
"match"
],
"track" : "Alignments",
"category" : "Alignments",
"class" : "feature4",
"key" : "Example alignments",
"hooks": {
"modify": "function( track, feature, div ) { div.style.height = (Math.random()*10+8)+'px'; div.style.backgroundColor = ['green','blue','red','orange','purple'][Math.round(Math.random()*5)];}"
},
"menuTemplate" : [
{
"label" : "Item with submenu",
# hello this is a comment
"children" : [
{
"label" : "Check gene on databases",
"children" : [
{
"label" : "Query trin for {name}",
"iconClass" : "dijitIconBookmark",
"action": "newWindow",
"url" : "http://wiki.trin.org.au/{name}-{start}-{end}"
},
{
"label" : "Query example.com for {name}",
"iconClass" : "dijitIconSearch",
"url" : "http://example.com/{name}-{start}-{end}"
}
]
},
{ "label" : "2nd child of demo" },
{ "label" : "3rd child: this is a track" }
]
},
{
"label" : "Open example.com in an iframe popup",
"title" : "The magnificent example.com (feature {name})",
"iconClass" : "dijitIconDatabase",
"action": "iframeDialog",
"url" : "http://www.example.com?featurename={name}"
},
{
"label" : "Open popup with XHR HTML snippet (btw this is feature {name})",
"title": "function(track,feature,div) { return 'Random XHR HTML '+Math.random()+' title!'; }",
"iconClass" : "dijitIconDatabase",
"action": "xhrDialog",
"url" : "sample_data/test_snippet.html?featurename={name}:{start}-{end}"
},
{
"label" : "Open popup with content snippet (btw this is feature {name})",
"title": "function(track,feature,div) { return 'Random content snippet '+Math.random()+' title!'; }",
"iconClass" : "dijitIconDatabase",
"action": "contentDialog",
"content" : "function(track,feature,div) { return '
\<h2\>{name}\<\/h2\> This is some test content about feature {name}! This message brought to you by the number \<span style=\"font-size: 300%\"\>'+Math.round(Math.random()\*100)+'\<\/span\>.
},
{
"label" : "function(track,feature,div) { return 'Run a JS callback '+Math.random()+' title!'; }",
"iconClass" : "dijitIconDatabase",
"action": "function( evt ){ alert('Hi there! Ran the callback on feature '+this.feature.get('name')); }"
},
{
"label": "Create a url with a callback",
"action": "iframeDialog",
"iconClass": "dijitIconDatabase",
"title": "Create a url with a callback",
"url": "function(track,feature) { return
'http://www.example.com?refseq='+track.refSeq.name
+'&featurename='+feature.get('name')+'&start='+feature.get('start')+'&end='+feature.get('end'); }"
},
]
}
This configuration results in a context menu like the one pictured below. For details on what each of the options supported by menu items does, see Click Configuration Options.
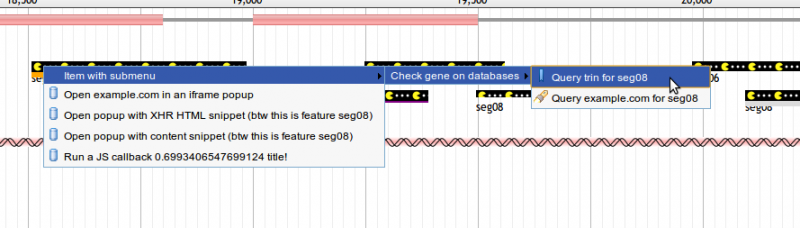
To add a separator, put the following item in your menuTemplate
{ type: 'dijit/MenuSeparator' }
Note that you can keep the default right-click menu items in JBrowse by just setting "blank" placeholders in the menuTemplate.
"menuTemplate" : [
{
"label" : "View details",
},
{
"label" : "Highlight this gene",
},
{
"label" : "Open example.com in an iframe popup",
"title" : "The magnificent example.com (feature{name})",
"iconClass" : "dijitIconDatabase",
"action": "iframeDialog",
"url" : "http://www.example.com?featurename={name}"
}
]
Alternatively, if you are using tracks.conf format, you can build a menuTemplate similar to the above configuration using the following:
menuTemplate+=json:{"label": "View details"}
menuTemplate+=json:{"label": "Highlight this gene"}
menuTemplate+=json:{"label": "Open example.com in an iframe popup", "iconClass" : "dijitIconDatabase","action": "iframeDialog","url" : "http://www.example.com?featurename={name}"}
This results in a context menu like the one pictured below.

Note: You'll note in the above that "placeholder" menu items are put in place to prevent the default "View details" from being overwritten. There are some caveats with regards to these placeholder items. You can't rearrange them or choose which ones you want. You can only overwrite them. Custom items will overwrite the default ones one-by-one.
A click action (left-click on a feature or on an item in a context menu) can be configured to do nearly anything. It can be configured with a string JavaScript callback, like:
"function( track, feature, featureDiv ) { alert('Run any JavaScript you want here!'); }"
Or a structure containing options like:
{
"iconClass" : "dijitIconDatabase",
"action" : "iframeDialog",
"url" : "http://www.ncbi.nlm.nih.gov/gquery/?term={name}",
"label" : "Search for {name} at NCBI",
"title" : "function(track,feature,div) { return 'Searching for '+feature.get('name')+' at NCBI'; }"
}
The available options for a click action are:
-
iconClass: Used only for click actions in context menus. Usually, you will want to specify one of the Dijit icon classes here. Although they are not well documented, a list of available icon classes can be seen at https://github.com/dojo/dijit/blob/1.7.2/icons/commonIcons.css.
-
action: Either a JavaScript function to run in response to the click (e.g. "function(){..}"), or one of the following special strings:
-
"iframeDialog" - the default - causes the given url to be opened in a popup dialog box within JBrowse, in an
iframeelement. -
"newWindow" - causes the given url to be opened in a new browser window.
-
"navigateTo" - added in JBrowse 1.10.8, opens the given url in the same browser window, navigating the user away from JBrowse.
-
"contentDialog" - causes the JavaScript string or callback set in the content option to be displayed in the dialog box.
-
"defaultDialog" - Performs the normal popup action. See JBrowse_Configuration_Guide#Customizing_Mouse-over_behavior for an example of when this is useful.
-
"xhrDialog" - causes the given url to be opened in a popup dialog, containing the HTML fetched from the given url option. The difference between "iframeDialog" and "xhrDialog" is that an iframeDialog's URL should point to a complete web page, while an xhrDialog's URL should point to a URL on the same server (or that supports CORS) that contains just a snippet of HTML (not a complete web page). For those familiar with GBrowse, the xhrDialog is similar to GBrowse popup balloons that use a url:... target, while the contentDialog is similar to a GBrowse popup balloon with a normal target. GBrowse does not have an equivalent of the iframeDialog.]
-
"Javascript callback" - If you use a javascript callback for the action parameter, then the function signature will be function(clickEvent) { ... } and the clickEvent doesn't contain particularly useful info, but you can access the feature object using this.feature.get('name'); for example.
-
content: string (interpolated with feature fields) or JS callback that returns either a string or (beginning in 1.10.0) a dojo/Deferred object that will resolve to a string. Used only by a contentDialog.
-
url: URL used by newWindow, xhrDialog, or iframeDialog actions.
-
label: descriptive label for the link. In a right-click context menu, this will be the text in the menu item. In a onClick section, it will be the mouse-over description too. See JBrowse_Configuration_Guide#Customizing_Mouse-over_behavior for details on the mouse-over behavior.
-
title: title used for the popup window
JBrowse feature tracks, and individual JBrowse features, can be customized using JavaScript functions you write yourself. These functions are called every time a feature in a track is drawn, and allow you to customize virtually anything about the feature's display. What's more, all of the feature's data is accessible to your customization function, so you can even customize individual features' looks based on their data.
As of JBrowse 1.3.0, feature callbacks are added by directly editing your trackList.json file with a text editor. Unfortunately, due to the limitations of the JSON format currently used for JBrowse configuration, the function must appear as a quoted (and JSON-escaped) string, on a single line. You may use the .conf format for the ability to specify functions that span multiple lines.
Here is an example feature callback, in context in the trackList.json file, that can change a feature's background CSS property (which controls the feature's color) as a function of the feature's name. If the feature's name contains a number that is odd, it give the feature's HTML div element a red background. Otherwise, it gives it a blue background.
{
"style" : {
"className" : "feature2"
},
"key" : "Example Features",
"feature" : [
"remark"
],
"urlTemplate" : "tracks/ExampleFeatures/{refseq}/trackData.json",
"hooks": {
"modify": "function( track, f, fdiv ) { var nums = f.get('name').match(/\\d+/); if( nums && nums[0] % 2 ) { fdiv.style.background = 'red'; } else { fdiv.style.background = 'blue'; } }"
},
"compress" : 0,
"label" : "ExampleFeatures",
"type" : "FeatureTrack"
},
JBrowse has several track types that are designed for displaying alignment data, particularly from BAM and CRAM files. BAM and CRAM files used with JBrowse must be compressed and sorted by leftmost coordinate.
The JBrowse BAM parsing library makes extensive use of code from BioDalliance, while the CRAM support is based on the @gmod/cram npm module.
Introduced in JBrowse 1.8.0, Alignments2 tracks are designed to display alignment data, such as from BAM files. This track type shows basepair-level mismatches, insertions, deletions, and skipped regions between aligned reads and the reference, and highlights paired reads in red if their mates are missing.
Base mismatches are displayed based on the contents of the feature's MD field (containing a BAM MD mismatch string), and/or CIGAR field. If your BAM file does not contain MD tags, one common way to generate them is with the samtools calmd command.
Alignments2 is a faster implementation of the older Alignments track type that draws alignments using the HTML5 canvas. In the interest of speed, Alignments2 tracks do not display any text labels or descriptions alongside features, and do not draw arrowheads indicating strandedness, instead relying on color to denote the strand of alignments.
Alignments2 tracks support the same advanced clicking behavior as CanvasFeatures tracks, but does not support right-click menus.
The most basic Alignments2 track configuration in tracks.conf format is
[tracks.alignments]
urlTemplate=FM.01.new.sorted.chr11.bam
type=Alignments2
The most basic track configuration in trackList.json format is
{
"label": "alignments",
"urlTemplate": "FM.01.new.sorted.chr11.bam",
"type": "Alignments2",
"storeClass": "JBrowse/Store/SeqFeature/BAM"
}
Note that this uses several tricks, including automatically inferring track type to be JBrowse/View/Track/Alignments2 from just saying Alignments2 (it could have also said "type": "JBrowse/View/Track/Alignments2", and it also defaults to using the storeClass JBrowse/Store/SeqFeature/BAM, which reads BAM files. You can explicitly specify "storeClass": "JBrowse/Store/SeqFeature/BAM" for completeness. Not all track types infer the storeClass.
List of configuration options:
| Option | Value |
|---|---|
maxHeight |
Available in JBrowse 1.9.0 and later. Maximum displayed height of the track in pixels. Defaults to 1000. Features that would cause the track to grow taller than the maxHeight are not shown. If a section of a track has features that are not drawn because of a maxHeight constraint, a notification is displayed at the bottom of that track section. |
style→color |
HTML-style color for drawn alignments. By default, this varies with the alignment's strandedness, and whether its mate pair is missing, using the style→color_fwd_strand, style→color_rev_strand, and style→color_missing_mate variables. To gain complete control over the displayed color, you could set this to be a function callback. |
style→color_fwd_strand |
HTML-style color for alignments on the forward strand. Default #EC8B8B, which is a light red. |
style→color_rev_strand |
HTML-style color for alignments on the reverse strand. Default #898FD8, which is a light blue. |
style→color_missing_mate |
HTML-style color for alignments with a missing mate. Default #D11919, which is a dark red. |
style→height |
Height in pixels of each alignment. Default 7. |
style→marginBotton |
Number of pixels of vertical spacing to put on the bottom of each alignment. Default 1. |
style→showMismatches |
If true, draw mismatches (SNPs, insertions, deletions, skips) on the alignent. Default true. |
style→mismatchFont |
CSS string describing the font to use for labeling mismatches. Default "bold 10px Courier New,monospace". |
histograms.storeClass |
A store class for summary histograms used for the Alignments2 track. Usually JBrowse/Store/SeqFeature/BigWig. Can be used on any CanvasFeatures-type track but generally used in Alignments2 tracks |
histograms.urlTemplate |
Path to a histogram file (such as a BigWig) to be used for summary histograms used for the Alignments2 track. Can be used on any CanvasFeatures-type track but generally used in Alignments2 tracks |
histograms.color |
Color for the histograms e.g. "purple". Default is orange. Can be used on any CanvasFeatures-type track but generally used in Alignments2 tracks |
histograms.binsPerBlock |
"Granularity" of the bins in histogram. Default is 200 for Alignments2 tracks. Default is 25 on other CanvasFeatures type tracks. |
hideDuplicateReads |
Hide duplicate reads to the same location. Default: true |
hideQCFailingReads |
Hide QC failing reads that did not pass some aligner quality. Default: true |
hideSecondary |
Hide secondary reads which mapped to multiple locations. Default: true |
hideUnmapped |
Hide unmapped reads. Default: true |
hideMissingMatepairs |
If a read is missing a mate pair or paired-end match, hide the read. Default: false |
hideForwardStrand |
Hide all reads from the forward strand. Default: false |
hideReverseStrand |
Hide all reads from the reverse strand. Default: false |
useReverseTemplate |
Use an algorithm for reversing the template of paired-end reads so that they appear on the same strand. Default: false. Added in 1.11.6 |
useReverseTemplateOption |
Present a checkbox to the user for changing the "Use reverse template" option. Default: true. Added in 1.11.6 |
useXS |
Use an algorithm for only coloring reads when the XS tag indicates strandedness. Default: false. Added in 1.11.6 |
useXSOption |
Present a checkbox to the user for changing the "Use XS" option. Default: true. Added in 1.11.6 |
cacheMismatches |
Cache mismatch calculations so that long reads are faster to browser. Default: false. Added in 1.12.3 |
renderAlignments |
Add a text display of the BAM alignment on a single line in the View details popup. Default: false |
renderPrettyAlignments |
Add a text display of the BAM alignment using prettier "BLAST style" to the View details popup. Default: false |
Since JBrowse 1.11.3, there is a new coloring scheme for BAM files that allows for new coloring of paired end reads, such as a different coloring for unpaired reads and aberrant pairing split across chromosomes.
The coloring styles that can be configured for the Alignments2 track are as follows
| Option |
|---|
style→color_fwd_strand |
style→color_rev_strand |
style→color_fwd_missing_mate |
style→color_rev_missing_mate |
style→color_fwd_strand_not_proper |
style→color_rev_strand_not_proper |
style→color_fwd_diff_chr |
style→color_rev_diff_chr |
style→color_nostrand |
If this scheme is undesirable, the style->color variable can be overridden entirely as well, with a callback for example
Introduced in JBrowse 1.8.0, SNPCoverage tracks draw the coverage of alignment features along the genome, along with a graphical representation of base-mismatch (possible SNP) distribution, and tables showing frequencies for each mismatching base.
Like the other alignment tracks, base mismatches are displayed based on the contents of the feature's MD field (containing a BAM MD mismatch string).
Note: Since the SNPCoverage track dynamically calculates coverage and putative SNPs directly from alignment data, it is not recommended for use with very dense feature data, such as deep-coverage BAM files. For these types of files, it's recommended to pre-generate a BigWig file of the coverage and a VCF file of putative SNPs, and display those instead.
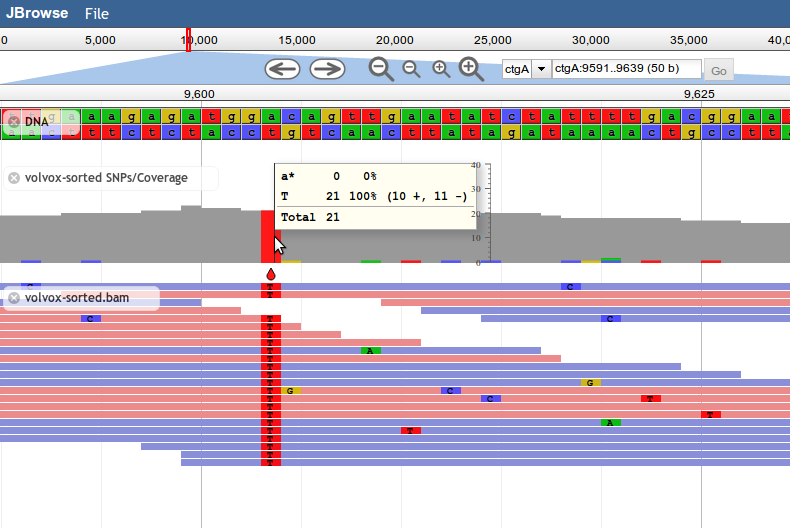
In your data/tracks.conf file:
[tracks.my-bam-coverage-track]
storeClass = JBrowse/Store/SeqFeature/BAM
urlTemplate = volvox-sorted.bam
type = JBrowse/View/Track/SNPCoverage
metadata.Description = SNP/Coverage view of volvox-sorted.bam, simulated resequencing alignments.
key = BAM - volvox-sorted SNPs/Coverage
Note that urlTemplate will refer to a file relative to the "data" directory that you are using.
Introduced in JBrowse 1.7.0, Alignments tracks are an HTML-based track type for alignment display. They display everything that Alignments2 do, and also can be configured with right-click menus and strand arrowheads.
They display everything that Alignments2 tracks do, plus they support the same configuration options as feature tracks, including advanced clicking behavior, feature modification callbacks, and so forth. The price of this additional capability is that Alignments tracks are significantly slower when used with dense data such as deep BAM alignments.
Alignments2 is recommended over Alignments for most users.
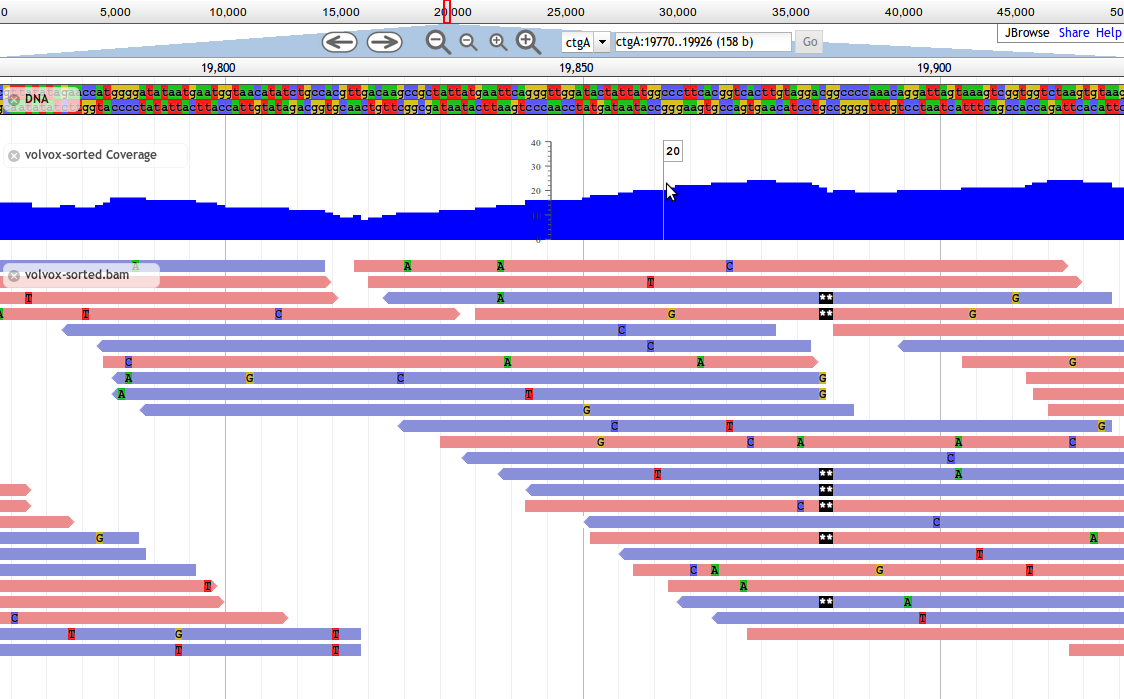
| Option | Value |
|---|---|
urlTemplate |
URL for the BAM file to display. |
baiUrlTemplate |
URL for the corresponding BAM index (BAI) file. If not set, this is assumed to be the same URL as urlTemplate with .bai appended. |
chunkSizeLimit |
Maximum size in bytes of BAM chunks that the browser will try to deal with. Default 5000000 (5 MiB). When this is exceeded, most tracks will display some kind of "Too much data" message. If you increase this, be careful. You could blow up your web browser. |
Note: you can also increase maxFeatureScreenDensity if you get the "Too much data to show; zoom in to see detail".
{
"storeClass" : "JBrowse/Store/SeqFeature/BAM",
"urlTemplate" : "../../raw/volvox/volvox-sorted.bam",
"label" : "volvox-sorted.bam",
"type" : "JBrowse/View/Track/Alignments2"
},
If you are using the Apache web server, please be aware that the module mime_magic can cause BAM files to be served incorrectly. Usually, the error in the web developer console will be something like "Not a BAM file". Some packaged versions of Apache, particularly on Red Hat or CentOS-based systems, are configured with this module turned on by default. We recommend you deactivate this Apache module for the server or directory used to serve JBrowse files. If you do not want to deactivate this module for the entire server, try adding this line to your HTTPD config or .htaccess file:
AddType application/octet-stream .bam .bami .bai
Introduced in JBrowse 1.5.0, Wiggle tracks require that the user's browser support HTML <canvas> elements. BigWig support requires a web browser with support for HTML5 typed arrays.
Beginning with JBrowse 1.7.0, there are two available subtypes of Wiggle tracks, Wiggle/XYPlot and Wiggle/Density. The XYPlot wiggle track displays quantitative data as an x/y plot, and the Density displays the data as varying intensities of color.
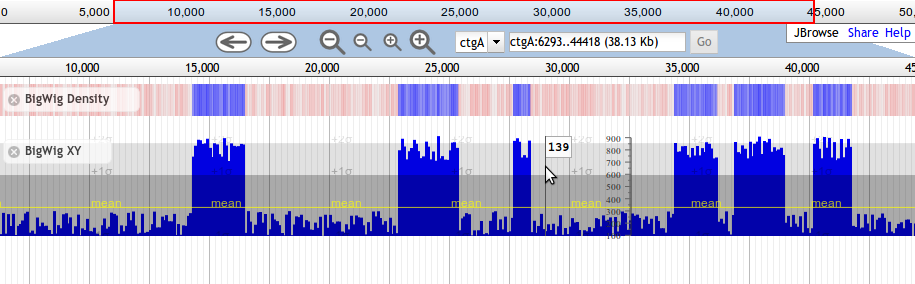
Here is an example track configuration stanza for a Wiggle XY-plot track displaying data directly from a BigWig file. Note that the URL in urlTemplate is relative to the directory where the configuration file is located.
{
"label" : "rnaseq",
"key" : "RNA-Seq Coverage",
"storeClass" : "JBrowse/Store/SeqFeature/BigWig",
"urlTemplate" : "../tests/data/SL2.40_all_rna_seq.v1.bigwig",
"type" : "JBrowse/View/Track/Wiggle/XYPlot",
"variance_band" : true,
"min_score" : -1000,
"max_score" : 2000,
"style": {
"pos_color" : "#FFA600",
"neg_color" : "#005EFF",
"clip_marker_color" : "red",
"height" : 100
}
}
Note: numerical values do not appear in quotes.
Here is an example track configuration stanza for a Wiggle color-density track displaying data directly from a BigWig file. It will draw regions with data that is greater than the overall mean as progressively more intense blue, and data that is below the mean as progressively more intense red. Note that the URL in urlTemplate is relative to the directory where the configuration file is located. Also note that green and purple is an ugly color combination. ;-)
{
"label" : "rnaseq",
"key" : "RNA-Seq Coverage",
"storeClass" : "JBrowse/Store/SeqFeature/BigWig",
"urlTemplate" : "my_methylation_data.bw",
"type" : "JBrowse/View/Track/Wiggle/Density",
"bicolor_pivot" : "mean",
"style": {
"pos_color": "purple",
"neg_color": "green"
}
}
| Option | Value |
|---|---|
yScalePosition |
Position of the y-axis scale indicator when the track is zoomed far enough out that density histograms are displayed. Can be "left", "right", or "center". Defaults to "center". |
origin_color |
Added in JBrowse 1.7.1, sets the color of the graph origin line in wiggle XY plots. If set to 'none', turns the origin line off. |
bg_color |
Added in JBrowse 1.7.0, sets the background color for each point at which data is present. Wiggle/Density tracks blend the background color with the pos_color or neg_color to select a color for the region, whereas Wiggle/XYPlot tracks draw the background color on the part of the graph for each data point that is not covered by the pos_color or neg_color. Defaults to transparent for XYPlots, and light gray for Density tracks. |
scale |
linear|log, default linear Graphing scale, either linear or logarithmic. |
min_score |
Number. The minimum value to be graphed. Calculated according to autoscale if not provided. |
max_score |
Number. The maximum value to be graphed. Calculated according to autoscale if not provided. |
autoscale |
local|clipped_global|global|z_score If one or more of min_score and max_score options are absent, then these values will be calculated automatically. The autoscale option controls how the calculation is done. A value of local (only available and set as the default in JBrowse 1.9.0 and above) sets the display scale to fit the range of the data being displayed in the current viewing window. A value of global will use global statistics (if available) for the entire quantitative dataset to find min_score and max_score values. z_score will use either ±z_score_bound if it is set, or will use ±4 otherwise. clipped_global is similar to global, except the bounds will be limited to ±z_score_bound, or ±4 if z_score_bound is not set. |
variance_band |
1 or 0 If 1, draw a yellow line showing the mean, and two shaded bands showing ±1 and ±2 standard deviations from the mean. |
z_score_bound |
for z-score based graphs, the bounds to use. |
data_offset |
number, default zero. If set, will offset the data display by the given value. For example, a data_offset of -100 would make a data value of 123 be displayed as 23, and a data_offset of 100 would make 123 be displayed as 223. |
bicolor_pivot |
"mean"|"zero"|(num) Where to change from pos_color to neg_color when drawing bicolor plots. Can be "mean", "zero", or a numeric value. Default 0. |
style→pos_color |
CSS color, default "blue". When drawing bicolor plots, the fill color to use for values that are above the pivot point. Can be a callback returning a color as well. |
style→neg_color |
CSS color, default "red". When drawing bicolor plots, the fill color to use for values that are below the pivot point. Can be a callback returning a color as well. |
disable_clip_markers |
boolean, default false. If true, disables clip markers, which are 2-pixel colored regions at the edge of the graph that indicate when the data value lies outside the displayed range. |
style→clip_marker_color |
CSS color, defaults to neg_color when in the positive bicolor regime (see bicolor_pivot) and pos_color in the negative bicolor regime. |
style→height |
Height, in pixels, of the track. Defaults to 100 for XYPlot tracks, and 32 for Density tracks. |
scoreType |
The scoreType to be used at the summary level. Options: maxScore, avgScore, score, minScore. Default: score is the backwards compatible default which is an average score when zoomed out, max score when zoomed in. maxScore is max score zoomed out and max score zoomed in. avgScore is both average score zoomed in and average score zoomed out. avgScore added in 1.12.0. maxScore/minScore added in 1.11.6. |
logScaleOption |
Add or remove the "Log scale" checkbox for the track menu. Default: true. Added in 1.11.6. |
noFill |
Draw the bigwig track as a "scatterplot" by not filling in the boxes. Default: false. Added in 1.12.3 |
At least one user has reported that BigWig files generated with older versions of Jim Kent's wigToBigWig tool are not compatible with JBrowse. If you encounter difficulties opening a BigWig file, try regenerating it with a more recent version of wigToBigWig, and reporting the problem to the mailing list.
Beginning in JBrowse 1.9.0, JBrowse can display feature data directly from VCF files, and has an HTMLVariants track type that is optimized for displaying the potentially large amounts of detailed data that go with each variant.
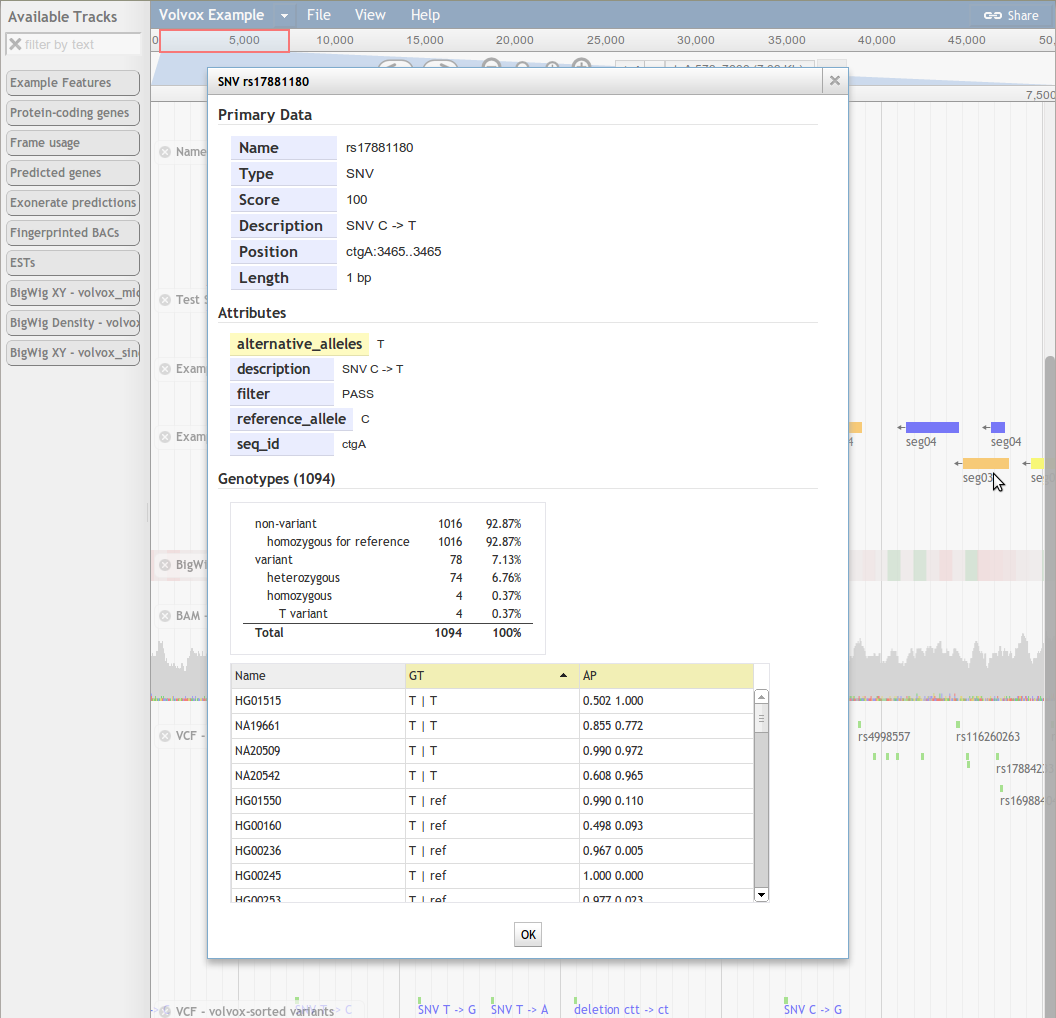
VCF files used with the VCFTabix must be compressed with bgzip and indexed with tabix, both of which are part of the samtools package. This is usually done with commands like:
bgzip my.vcf
tabix -p vcf my.vcf.gz
Here is an example track configuration stanza for a variant track displaying data directly from a VCF file. Note that the URL in urlTemplate is relative to the directory where the configuration file is located. Note that tbiUrlTemplate can also be used if your tbi file is named anything other than the urlTemplate with .tbi added to the end.
{
"label" : "mysnps",
"key" : "SNPs from VCF",
"storeClass" : "JBrowse/Store/SeqFeature/VCFTabix",
"urlTemplate" : "../vcf_files/SL2.40_all_rna_seq.v1.vcf.gz",
"type" : "JBrowse/View/Track/HTMLVariants"
}
Alternatively, if you are using the tracks.conf format, then a similar example would look like the following
[ tracks.myvcf ]
# settings for what data is shown in the track
storeClass = JBrowse/Store/SeqFeature/VCFTabix
urlTemplate = ../vcf_files/SL2.40_all_rna_seq.v1.vcf.gz
# settings for how the track looks
category = VCF
type = JBrowse/View/Track/CanvasVariants
key = SNPs from VCF
The two variables hideNotFilterPass or hideFilterPass can be used to define whether to filter some variants by default. For example adding hideNotFilterPass: 1 will show the variants that passed all filters by default (i.e. it hides all the features that didn't pass all the filters)
"hideNotFilterPass": 1
Introduced in JBrowse 1.7.0, feature coverage tracks show a dynamically-computed XY-plot of the depth of coverage of features across a genome. One good use of this track type is to provide a quick coverage plot directly from a BAM file. However, since this track calculates coverage on the fly, it can be very slow when used with large regions or very deep coverage. In this case, it is recommended to generate a BigWig file containing the coverage data, and display it with a Wiggle/XYPlot or Wiggle/Density track.
Feature coverage tracks are a special type of Wiggle/XYPlot tracks, so the same configuration options apply. There is an additional caveat, however: this kind of track requires the min_score and max_score variables in order to set the Y-axis scaling, since these cannot be quickly determined from raw BAM or other feature data.
Note: The SNPCoverage track and FeatureCoverage tracks are very similar, except the SNPCoverage track (in addition to showing SNPs) also has the extra ability to filter the supplementary/secondary reads, and other reads, so they may appear to report different coverages by default.
{
"storeClass" : "JBrowse/Store/SeqFeature/BAM",
"urlTemplate" : "../../raw/volvox/volvox-sorted.bam",
"label" : "volvox-sorted.bam_coverage",
"type" : "JBrowse/View/Track/FeatureCoverage",
"min_score" : 0,
"max_score" : 35,
"key" : "volvox-sorted Coverage"
}
The Sequence track added support for using a specified codon table or partial codon table, with start or stop codons highlighted
| Option | Value |
|---|---|
codonTable |
Specify a codon table or partial codon table. See http://www.ncbi.nlm.nih.gov/Taxonomy/Utils/wprintgc.cgi. Example "codonTable": { "AAA": "N" }. Available since 1.11.6 |
codonStarts |
Specify a set of start codons. See http://www.ncbi.nlm.nih.gov/Taxonomy/Utils/wprintgc.cgi. Example "codonStarts": [ "AAA" ]. Available since 1.12.0 |
codonStops |
Specify a set of stop codons. See http://www.ncbi.nlm.nih.gov/Taxonomy/Utils/wprintgc.cgi. Example "codonStops": [ "AAA" ]. Available since 1.12.0 |
useAsRefSeqStore |
Make a given track specifically identified as a refseq store, and JBrowse will then use the store class specified on your track to retrieve the data for the FASTA in "View details" popups, etc. |
Note that the colors of nucleotide bases rendered is controlled via CSS, so you can add custom CSS to a plugin or edit the jbrowse CSS to override it. The classes are
base_n,base_a,base_g,base_t,base_c
The amino acid track highlights can also be controlled via CSS. They can be either the letter "aminoAcid_m" for example or the "aminoAcid_start", "aminoAcid_stop" classes
.translatedSequence td.aminoAcid_start
.translatedSequence td.aminoAcid_m
.translatedSequence td.aminoAcid_k
JBrowse supports tracks based on pre-generated PNG or JPEG images that are tiled along the reference sequence. Currently, JBrowse ships with two different image track generators: wig-to-json.pl, which generates images showing simple quantitative (wiggle) data, and draw-basepair-track.pl, which draws arcs to show the base pairing structure of RNAs.
Using a wiggle file, this script creates a single Image track that displays data from the wiggle file. Beginning with JBrowse 1.5, this is no longer the recommended method of displaying wiggle data: it has largely been replaced by the direct-access BigWig data store coupled with the next-generation Wiggle track type. See Wiggle Tracks.
In wiggle data, a numeric value is associated with each nucleotide position in the reference sequence. This is represented in JBrowse as a track that looks like a histogram, where the horizontal axis is for each nucleotide position, and the vertical axis is for the number associated with that position. The vertical axis currently does not have a scale; rather, the heights for each position are relative to each other.
Special dependencies: libpng
In order to use wig-to-json.pl, the code for wig2png must be compiled. Normally, this is done automatically by setup.sh but it can be done manually if necessary. See the Quick Start Tutorial packaged with JBrowse for details.
bin/wig-to-json.pl --wig <wig file> --tracklabel <track name> [options]
Hint: If you are using this type of track to plot a measure of a prediction's quality, where the range of possible quality scores is from some lowerbound to some upperbound (for instance, between 0 and 1), you can specify these bounds with the max and min options.

| Option | Value |
|---|---|
| wig | The name of the wig file that will be used. This option must be specified. |
| tracklabel | The internal name that JBrowse will give to this feature track. This option requires a value. |
| key | The external, human-readable label seen on the feature track when it is viewed in JBrowse. The value of key defaults to the value of tracklabel. |
| out | A path to the output directory (default is 'data' in the current directory). |
| tile | The directory where the tiles, or images corresponding to each zoom level of the track, are stored. Defaults to data/tiles. |
| bgcolor | The color of the track background. Specified as "RED,GREEN,BLUE" in base ten numbers between 0 and 255. Defaults to "255,255,255". |
| fgcolor | The color of the track foreground (i.e. the vertical bars of the wiggle track). Specified as "RED,GREEN,BLUE" in base ten numbers between 0 and 255. Defaults to "105,155,111". |
| width | The width in pixels of each tile. The default value is 2000. |
| height | The height in pixels of each tile. Changing this parameter will cause a corresponding change in the top-to-bottom height of the track in JBrowse. The default value is 100. |
| min | The lowerbound to use for the track. By default, this is the lowest value in the wiggle file. |
| max | The upperbound to use for the track. By default, this will be the highest value in the wiggle file. |
You need libpng and a C++ compiler; you can get these with Homebrew, MacPorts, or Fink.
Once you have those installed, you need to compile the JBrowse wiggle-processing program. JBrowse includes a makefile to do this, but you may need to add the paths for libpng and png.h to your compiler's library and include paths. For example, if libpng is in /usr/X11, you can run configure like this:
./configure CXXFLAGS=-I/usr/X11/include LDFLAGS=-L/usr/X11/lib
Depending on which OS X you have (and whether you're using MacPorts/fink/etc.), libpng might be somewhere other than /usr/X11, like /opt/local for example. To find where libpng is, try locate libpng.
Be sure to install the libpng\#\# and libpng\#\#-dev packages, where \#\# is a number such as 12 or 15, depending on the version of Ubuntu.
Be sure to install the libpng and libpng-devel packages.
This script inputs a single base pairing track into JBrowse. A base pairing track is a distinctive track type that represents base pairing between nucleotides as arcs. In addition, it is intended to demonstrate the Perl API for writing your own image track generators.
bin/draw-basepair-track.pl --gff <gff file> --tracklabel <track name> [options]

| Option | Value |
|---|---|
| gff | The name of the gff file that will be used. This option must be specified. |
| tracklabel | The internal name that JBrowse will give to this feature track. This option requires a value. |
| key | The external, human-readable label seen on the feature track when it is viewed in JBrowse. The value of key defaults to the value of tracklabel. |
| out | A path to the output directory (default is 'data' in the current directory). |
| tile | The directory where the tiles, or images corresponding to each zoom level of the track, are stored. Defaults to data/tiles. |
| bgcolor | The color of the track background. Specified as "RED,GREEN,BLUE" in base ten numbers between 0 and 255. Defaults to "255,255,255". |
| fgcolor | The color of the track foreground (i.e. the base pairing arcs). Specified as "RED,GREEN,BLUE" in base ten numbers between 0 and 255. Defaults to "0,255,0". |
| width | The width in pixels of each tile. The default value is 2000. |
| height | The height in pixels of each tile. Changing this parameter will cause a corresponding change in the top-to-bottom height of the track in JBrowse. The default value is 100. |
| thickness | The thickness of the base pairing arcs in the track. The default value is 2. |
| nolinks | Disables use of file system links to compress duplicate image files. |
The JBrowse search box auto-completes the names of features and reference sequences that are typed into it. After loading all feature and reference sequence data into a JBrowse instance (with prepare-refseqs.pl, flatfile-to-json.pl, etc.), generate-names.pl must be run to build the indexes used for name searching and autocompletion.
Several settings are available to customize the behavior of autocompletion. Most users will not need to configure any of these variables.
| Option | Value |
|---|---|
autocomplete→stopPrefixes |
Array of word-prefixes for which autocomplete will be disabled. For example, a value of ['foo'] will prevent autocompletion when the user as typed 'f', 'fo', or 'foo', but autocompletion will resume when the user types any additional characters. |
autocomplete→resultLimit |
Maximum number of autocompletion results to display. Defaults to 15. |
autocomplete→tooManyMatchesMessage |
Message displayed in the autocompletion dropdown when more than autocomplete→resultLimit matches are displayed. Defaults to 'too many matches to display'. |
This script builds indexes of features by label (the visible name below a feature in JBrowse) and/or by alias (a secondary name that is not visible in the web browser, but may be present in the JSON used by JBrowse).
To search for a term, type it in the autocompleting text box at the top of the JBrowse window.
Basic syntax:
bin/generate-names.pl [options]
Note that generate-names.pl does not require any arguments. However, some options are available:
| Option | Value |
|---|---|
| --out | A path to the output directory (default is 'data/' in the current directory). |
| --verbose | This setting causes information about the division of nodes into chunks to be printed to the screen. |
| --hashBits | Sets the number of bits, or the filename length. If you get the error "Error reading from name store", try manually setting --hashBits to 16. |
| --completionLimit | Defines the number of letters used in the auto-completion. Example: --completionLimit 0 to disable autocompletion. |
| --incremental | Incrementally index new tracks. Fixed in version 1.11.6. |
| --tracks | A comma separated list of tracks to index. Can be combined with other options such as --incremental and --completionLimit to only provide autocompletion on certain tracks. |
| --workdir | Output the temporary files for names generation to a specific directory. Fixed in 1.11.6. |
View bin/generate-names.pl --help for more options. Note that if you are getting 404 errors for names/root.json then JBrowse is falling back to the legacy names store (and failing) so it is likely that you need to retry generate-names.
Note: by defeault, the Name, ID, and Alias fields are indexed by generate-names.pl because those are what are specified to be the "names" of the features when you run flatfile-to-json.pl, however, if you run flatfile-to-json.pl with --nameAttributes "name,id,alias,gene_id" for example, then it will also load the "gene_id" field as a name, and then you can re-run generate-names.pl and the gene_id can be searched for.
JBrowse has a script to remove individual tracks: remove-track.pl. Run it with the --help option to see a comprehensive usage message:
bin/remove-track.pl --help
JBrowse works with HTTP Basic, HTTP Digest, and cookie (session) authentication methods, relying on the native support for them in browsers.
For cookie-based authentication methods, the session cookie should be set by another page before the user launches JBrowse.
Provided you have a LDAP authentication server already available it is relatively easy to configure nginx to require users to login and optionally be members of particular groups.
This approach is designed to block access to all of JBrowse until authenticated and is not suitable for excluding sub-sets of tracks.
The following block lists the installation method for the module and dependancies with versions available at time of writing:
sudo apt-get install libldap2-dev
sudo apt-get install build-essential
sudo apt-get install libcurl4-openssl-dev
mkdir ldap_test
cd ldap_test/
wget http://nginx.org/download/nginx-1.10.1.tar.gz
tar zxf nginx-1.10.1.tar.gz
wget http://zlib.net/zlib-1.2.8.tar.gz
tar zxf zlib-1.2.8.tar.gz
wget ftp://ftp.csx.cam.ac.uk/pub/software/programming/pcre/pcre-8.37.tar.gz
tar zxf pcre-8.37.tar.gz
wget https://github.com/kvspb/nginx-auth-ldap/archive/master.zip
unzip master.zip
rm *.zip *.gz
cd nginx-1.10.1/
./configure --prefix=/jbrowse/nginx_ldap --with-zlib=../zlib-1.2.8 --with-pcre=../pcre-8.37 --with-http_ssl_module --add-module=../nginx-auth-ldap-master
make install
pcre2 is not compatible, you must use pcre-X.XX
The next block shows an example configuration that would be added to the 'http' section of 'nginx.conf'
http {
...
# for any user who successfully authenticates against LDAP
ldap_server shared_site {
# user search base.
url "ldap://ldap-ro.internal.example.ac.uk/dc=example,dc=ac,dc=uk?uid?sub?objectClass=person";
# bind as
binddn "uid=WEBSERVER_USER,ou=people,dc=example,dc=ac,dc=uk";
# bind pw
binddn_passwd "WEBSERVER_USER_PW";
# group attribute name which contains member object
group_attribute member;
# search for full DN in member object
group_attribute_is_dn on;
# matching algorithm (any / all)
satisfy any;
require valid_user;
}
# just our sub team
ldap_server team_only
{
# exactly the same as above but adding:
# list of allowed groups
require group "CN=mygroup,OU=group,DC=example,DC=ac,DC=uk";
}
You may need to use 'ldapsearch' or speak to your admins for help getting the settings correct.
Once this is in place you can then limit the accessible locations by adding to the 'server' section:
server {
...
# this is open access
location / {
root html;
index index.html index.htm;
}
# these require authentication
location /shared_site {
auth_ldap "Restricted access cancer members only";
auth_ldap_servers shared_site;
}
location /team_only {
auth_ldap "Restricted access cgppc members only";
auth_ldap_servers team_only;
}
...
}
If you place the 'auth_ldap*' directives before the location sections then you restrict all areas.
This was pieced together from the following pages:
Because of JavaScript's same-origin security policy, if data files shown in JBrowse do not reside on the same server as JBrowse, the web server software that serves them must be configured to allow access to them via Cross-origin resource sharing (CORS).
Example wide-open CORS configuration for Apache:
<IfModule mod_headers.c>
Header onsuccess set Access-Control-Allow-Origin *
Header onsuccess set Access-Control-Allow-Headers X-Requested-With,Range
</IfModule>
The CORS Range header is needed to support loading byte-range pieces of BAM, VCF, and other large files from Remote URLs. If you receive an error that says "Unable to fetch " using Remote URLs, then check to make sure that the proper CORS settings are enabled on the server that you are loading the file from.
Starting with JBrowse 1.3.0, server-side data-formatting scripts support a --compress option to compress (gzip) feature and sequence data to conserve server disk space and reduce server CPU load even further. Using this option requires some additional web server configuration.
For Apache:
-
AllowOverride FileInfo(orAllowOverride All) must be set for the JBrowse data directories in order to use the.htaccessfiles generated by the formatting scripts. -
mod_headersmust be installed and enabled, and if the web server is usingmod_gzipormod_deflate,mod_setenvifmust also be installed and enabled.
For Apache: A configuration snippet like the following should be included in the configuration:
<IfModule mod_gzip.c>
mod_gzip_item_exclude "(\.jsonz|\.txtz)"
</IfModule>
<IfModule setenvif.c>
SetEnvIf Request_URI "(\.jsonz|\.txtz)" no-gzip dont-vary
</IfModule>
<IfModule mod_headers.c>
<FilesMatch "(\.jsonz|\.txtz)">
Header onsuccess set Content-Encoding gzip
</FilesMatch>
</IfModule>
For nginx: A configuration snippet like the following should be included in the configuration:
location ~* "\.(json|txt)z$" {
add_header Content-Encoding gzip;
gzip off;
types { application/json jsonz; }
}
List of default available JBrowse URL params, more details below
| Option | Value |
|---|---|
| data | Set the data directory being used. If you have multiple data directories, then you can select the active one using http://mysite/jbrowse/?data=rice or http://mysite/jbrowse/?loc=soybean (also see Configuration_loading_details and Multiple_genomes) |
| loc | A location, specified as "chr:start..end" or gene identifier (which is searched from a name store generated by generate-names.pl) e.g. http://mysite/jbrowse/?loc=chr1:0..1000 |
| highres | A configuration for highResolutionMode. Available since JBrowse 1.12.3. Can be auto,disabled, or a specific scaling ratio such as 2,3 |
| fullviewlink,menu,nav,overview,tracklist | Boolean to add or remove different components of the UI. Can be set to false to remove |
| tracks | Set a list of active tracks, comma separated list of *track labels* e.g. http://mysite/jbrowse/?tracks=DNA,GeneTrack |
| highlight | Set a highlighted region, same format as loc param |
| addFeatures,addStores,addTracks,addBookmarks | A JSON configuration for extra track info. See below for detailed descriptions |
JBrowse's default index.html file provides a number of options for changing the current view in the browser by adding query parameters to the URL. Developers' note: these URL parameters are handled only in the default index.html that ships with JBrowse; the JBrowse code itself does not pay attention to URL parameters.
Basic syntax:
http://<server>/<path to jbrowse>?loc=<location string>&tracks=<tracks to show>
Optional relative (or absolute) URL of the base directory from which JBrowse will load data. Defaults to 'data'.
The initial genomic position which will be visible in the viewing field. Possible input strings are:
"Chromosome"+":"+ start point + ".." + end point
A chromosome name/ID followed by “:”, starting position, “..” and end position of the genome to be viewed in the browser is used as an input. Chromosome ID can be either a string or a mix of string and numbers. “CHR” to indicate chromosome may or may not be used. Strings are not case-sensitive. If the chromosome ID is found in the database reference sequence (RefSeq), the chromosome will be shown from the starting position to the end position given in URL.
example) ctgA:100..200
Chromosome ctgA will be displayed from position 100 to 200.
OR start point + ".." + end point
A string of numerical value, “..” and another numerical value is given with the loc option. JBrowse navigates through the currently selected chromosome from the first numerical value, start point, to the second numerical value, end point.
example) 200..600
OR center base
If only one numerical value is given as an input, JBrowse treats the input as the center position. Then an arbitrary region of the currently selected gene is displayed in the viewing field with the given input position as basepair position on which to center the view.
OR feature name/ID
If a string or a mix of string and numbers are entered as an input, JBrowser treats the input as a feature name/ID of a gene. If the ID exists in the database RefSeq, JBrowser displays an arbitrary region of the feature from the the position 0, starting position of the gene, to a certain end point.
example) ctgA
parameters are comma-delimited strings containing track names, each of which should correspond to the "label" element of the track information dictionaries that are currently viewed in the viewing field. Names for the tracks can be found in data/trackInfo.js in jbrowse-1.2.1 folder.
Example:
tracks=DNA,knownGene,ccdsGene,snp131,pgWatson,simpleRepeat
location string to highlight. Example:
highlight=ctgA:100..200
Available in JBrowse 1.10.0 and above. This variable accepts feature data in JSON format in the form:
[{ "seq_id":"ctgA", "start": 123, "end": 456, "name": "MyBLASTHit"},...}]
which, when URI-escaped and put in the query string, looks like:
addFeatures=%5B%7B%20%22seq_id%22%3A%22ctgA%22%2C%20%22start%22%3A%20123%2C%20%22end%22%3A%20456%2C%20%22name%22%3A%20%22MyBLASTHit%22%7D%5D
in dot-notation, the second number is the order of the feature object in the JSONArray:
addFeatures.<featurenumber>.<values>
e.g.
addFeatures.1.seq_id=ctgA&addFeatures.1.start=123&addFeatures.1.start=456&addFeatures.1.name=MyBLASTHit&addFeatures.2.seq_id=...
Developers integrating JBrowse into larger project may find this feature useful for displaying results from other non-JavaScript-based applications (such as web BLAST tools) in JBrowse.
Features added to JBrowse in this way are available in a special data store named url, which can be specified in a track configuration by adding "store":"url".
This variable accepts track configurations in JSON format to be added to the tracks already in the JBrowse configuration, in the form:
[{"label":"mytrack","type":"JBrowse/View/Track/HTMLFeatures","store":"myUniqueStore"}]
which, when URI-escaped and put in the query string, looks like:
addTracks=%5B%7B%22label%22%3A%22mytrack%22%2C%22type%22%3A%22JBrowse%2FView%2FTrack%2FHTMLFeatures%22%2C%22store%22%3A%22myUniqueStore%22%7D%5D
using dot notation. See release-notes.txt for more details:
addTracks.myUniqueStore.label=mytrack&addTracks.myUniqueStore.myUniqueStore.type=JBrowse/View/Track/HTMLFeatures
If you are not adding a store, make the store name 'none' (addTracks.none.label).
This variable accepts store configurations in JSON format to be added to the stores already in the JBrowse configuration, in the form:
{ "uniqueStoreName": {
"type":"JBrowse/Store/SeqFeature/GFF3",
"urlTemplate": "url/of/my/file.gff3"
}
}
which, when URI-escaped and put in the query string, looks like:
addStores=%7B%22uniqueStoreName%22%3A%7B%22type%22%3A%22JBrowse%2FStore%2FSeqFeature%2FGFF3%22%2C%22urlTemplate%22%3A%22url%2Fof%2Fmy%2Ffile.gff3%22%7D%7D
using dot notation. See release-notes.txt for more details:
addStores.uniqueStoreName.type=JBrowse/Store/SeqFeature/GFF3&addStores.uniqueStoreName.urlTemplate=url/of/my/file.gff3
One example is to use addStores combined with addTracks to dynamically load a remote file. Thanks to vkrishna for the example
addStores={"url":{"type":"JBrowse/Store/SeqFeature/GFF3","urlTemplate":"http://host/genes.gff"}}&addTracks=[{"label":"genes","type":"JBrowse/View/Track/CanvasFeatures","store":"url"}]
Another example if you want to not provide the entire URL to the store (e.g. provide a path relative to data directory instead of http://host/genes.gff) is to use a combination of the baseUrl and the {dataRoot} variables. This example adds a VCF track:
addStores={"mystore":{"type":"JBrowse/Store/SeqFeature/VCFTabix","baseUrl":".","urlTemplate":"{dataRoot}/combined.vcf.gz"}}&addTracks=[{"label":"variants","type":"JBrowse/View/Track/CanvasVariants","store":"mystore"}]
In dot-notation:
addStores.mystore.type=JBrowse/Store/SeqFeature/VCFTabix&addStores.mystore.baseUrl=.&addStores.mystore.urlTemplate="{dataRoot}/combined.vcf.gz"&addTracks.mystore.label=variants&addTracks.mystore.type=Browse/View/Track/CanvasVariants
Thanks to biojiangke for inquiring about the above example.
Bookmarks are available in JBrowse starting in 1.12.2, which allows you to add static coloured overlays to specific regions of the genome.
This variable accepts track bookmarks (similar to highlights) in JSON format to be added to the tracks already in the JBrowse configuration, in the form:
[{"start":5500,"end":6000,"color": "rgba(190,50,50,0.5)","ref": "ctgB"}]
which, when URI-escaped and put in the query string, looks like:
addBookmarks=%5B%7B%22start%22%3A5500%2C%22end%22%3A6000%2C%22color%22%3A%20%22rgba(190%2C50%2C50%2C0.5)%22%2C%22ref%22%3A%20%22ctgB%22%7D%5D
using dot notation. See release-notes.txt for more details:
addBookmarks.start=5500&addBookmarks.end=6000&addBookmarks.color="rgba(190,50,50,0.5)"&addBookmarks.ref=ctgB
Bookmarks can also be manually added to the configuration, e.g. tracks.conf:
[ bookmarks ]
features+= json:{"start":5000,"end":5750,"color": "rgba(190,50,50,0.5)","ref": "ctgB"}
features+= json:{"start":5500,"end":6000,"color": "rgba(190,50,50,0.5)","ref": "ctgB"}
They can also be retrieved from a remote "bookmark API", which receives a query parameter "sequence" for the current refseq and returns data in the JSON format similar to above
bookmarkService=http://yourbookmarkservice.com/jbrowseBookmark
An example appears in the Volvox example, on ctgB.

JBrowse's included index.html file supports three URL query arguments that can turn off the JBrowse track list, navigation bar, and overview bar, respectively. When all three of these are turned off, the only thing visible are the displayed tracks themselves, and JBrowse could be said to be running in a kind of "embedded mode".
The three parameters used for this are nav, tracklist, menu and overview. If any of these are set to 0, that part of the JBrowse interface is hidden.
For example, you could put the embedded-mode JBrowse in an iframe, like this:
<html>
<head>
<title>
JBrowse Embedded
</title>
</head>
<body>
<h1>
Embedded Volvox JBrowse
</h1>
<div style="width: 400px; margin: 0 auto;">
<iframe style="border: 1px solid black" src="../../index.html?data=sample_data/json/volvox&tracklist=0&nav=0&overview=0&tracks=DNA%2CExampleFeatures%2CNameTest%2CMotifs%2CAlignments%2CGenes%2CReadingFrame%2CCDS%2CTranscript%2CClones%2CEST" width="300" height="300">
</iframe>
</div>
</body>
</html>
Note that if you are embedding with an iframe, it can be useful to keep the functionality that jbrowse has to update the URL bar in the parent frame. See this FAQ entry for details http://gmod.org/wiki/JBrowse_FAQ#How_can_I_get_jbrowse_to_update_the_URL_of_a_parent_page_when_jbrowse_is_inside_of_an_iframe
Starting with version 1.7.0, JBrowse users can export track data in a variety of formats for either specific regions, or for entire reference sequences. Export functionality can also be limited and disabled on a per-track basis using the configuration variables listed below.
Current supported export data formats are:
- FASTA (sequence tracks)
- GFF3 (all tracks)
- bed (feature and alignment tracks)
- bedGraph (wiggle tracks)
- Wiggle (wiggle tracks)
Each track in JBrowse that can export data supports the following configuration variables.
| Option | Value |
|---|---|
noExport |
If true, disable all data export functionality for this track. Default false. |
maxExportSpan |
Maximum size of the a region, in bp, that can be exported from this track. Default 500 Kb. |
maxExportFeatures |
Maximum number of features that can be exported from this track. If "Save track" is unexpectedly greyed out, inspect this setting. Default: 50000 |
maxFeatureSizeForUnderlyingRefSeq |
Maximum length of sequence to be displayed in the popup boxes. Default: 250kb |
Starting with version 1.10.0, JBrowse can display feature or quantitative data directly from a SPARQL endpoint. The SPARQL data adaptor can be used with any of the JBrowse track types.
To display annotations from a SPARQL endpoint, first write a SPARQL query that fetches features for a given reference sequence region, given the reference sequence name and the start and end coordinates of the region of interest, with one feature returned per output row. JBrowse will run this query every time it fetches features for a certain region. The reference sequence name, start, and end, are interpolated into the query at every occurrance of "{ref}", "{start}", or "{end}", respectively. This is the same variable interpolation syntax used in other parts of the JBrowse configuration.
Queries used with JBrowse can have any number of output columns, but are required to have at least 4: ?start, ?end, ?strand, and ?uniqueID (usually just the URI of the feature). If the data includes subfeatures, a ?parentUniqueID column can be added to the SPARQL query, and features will be attached as subfeatures to any feature in the query with that ?uniqueID. Any number of additional columns can be added, as well. Their contents will just be attached to each feature as attributes, which will be visible in the default feature detail dialog. If available, it's a good idea to add a ?name column, which would be the feature's displayed name, and maybe a ?description column, which can be a longer text description of the feature.
The example configuration below displays complete gene models (with locations represented using FALDO) contained in a SPARQL endpoint located at /sparql on the same server as JBrowse.
[tracks.genes]
label = genes
key = SPARQL Genes
storeClass = JBrowse/Store/SeqFeature/SPARQL
type = JBrowse/View/Track/CanvasFeatures
urlTemplate = /sparql
queryTemplate =
DEFINE sql:select-option "order"
prefix rdf: <http://www.w3.org/1999/02/22-rdf-syntax-ns#>
prefix rdfs: <http://www.w3.org/2000/01/rdf-schema#>
prefix xsd: <http://www.w3.org/2001/XMLSchema#>
prefix obo: <http://purl.obolibrary.org/obo/>
prefix faldo: <http://biohackathon.org/resource/faldo#>
prefix idorg: <http://rdf.identifiers.org/database/>
prefix insdc: <http://insdc.org/owl/>
select ?start,
?end,
IF( ?faldo_type = faldo:ForwardStrandPosition,
1,
IF( ?faldo_type = faldo:ReverseStrandPosition,
-1,
0
)
) as ?strand,
str(?obj_type_name) as ?type,
str(?label) as ?name,
str(?obj_name) as ?description,
?obj as ?uniqueID,
?parent as ?parentUniqueID
from <http://togogenome.org/refseq/>
from <http://togogenome.org/so/>
from <http://togogenome.org/faldo/>
where {
values ?faldo_type { faldo:ForwardStrandPosition faldo:ReverseStrandPosition faldo:BothStrandsPosition }
values ?refseq_label { "{ref}" }
#values ?obj_type { obo:SO_0000704 }
?obj obo:so_part_of ?parent . filter( ?obj_type = obo:SO_0000704 || ?parent != ?seq )
# on reference sequence
?obj obo:so_part_of+ ?seq .
?seq a ?seq_type.
?seq_type rdfs:label ?seq_type_label.
?seq rdfs:seeAlso ?refseq .
?refseq a idorg:RefSeq .
?refseq rdfs:label ?refseq_label .
# get faldo begin and end
?obj faldo:location ?faldo .
?faldo faldo:begin/rdf:type ?faldo_type .
?faldo faldo:begin/faldo:position ?start .
?faldo faldo:end/faldo:position ?end .
filter ( !(?start > {end} || ?end < {start}) )
# feature type
?obj rdf:type ?obj_type .
?obj_type rdfs:label ?obj_type_name .
optional {
?obj insdc:feature_locus_tag ?label .
}
# feature name is the feature product
optional {
?obj insdc:feature_product ?obj_name .
}
#optional {
# ?obj rdfs:seeAlso ?obj_seealso .
#}
}
By default, only "{ref}", "{start}", and "{end}" are available for interpolating into your query. However, starting with JBrowse 1.10.3, you can add additional variables in the configuration by including a variables key containing additional values. For example, you could add an "{organism_uri}" in your queryTemplate that was set from the variables stanza, which would look like:
{
"label": 'genes',
"key": "SPARQL Genes",
"storeClass": "JBrowse/Store/SeqFeature/SPARQL",
"type": 'JBrowse/View/Track/HTMLFeatures',
"urlTemplate": "/sparql",
"style": { "className": "transcript" },
"queryTemplate": "... {organism_uri} ...",
"variables": {
"organism_uri": "<http://my.organism.authority/tyrannosaurus_rex>"
}
}
The variable interpolation can also be used to refer to functions that are defined in external files (see Including external files and functions).
JBrowse supports two ways to add data that describes tracks (track metadata): it can either be embedded directly in the track's configuration stanza, or it can come from a separate file that JBrowse loads. Track metadata is shown both when a user clicks "About this track" from a track's menu, and in the faceted track selector if it is in use.
Each track configuration stanza can include a metadata item that contains items of data that describe the track. For example, to describe a BAM track containing alignments of RNA-seq reads from Volvox carteri under conditions of caffeine starvation, a track configuration might contain:
{
"storeClass" : "JBrowse/Store/SeqFeature/BAM",
"urlTemplate" : "../../raw/volvox/volvox-sorted.bam",
"style" : {
"className" : "alignment",
"arrowheadClass" : "arrowhead",
"labelScale" : 100
},
"label" : "volvox-sorted.bam",
"type" : "JBrowse/View/Track/Alignments",
"key" : "volvox-sorted.bam",
"metadata": {
"Description": "RNA-seq",
"Conditions": "caffeine starvation",
"Species": "Volvox carteri",
"Data Provider": "Robert Buels Institute for Example Data"
}
}
To add track metadata from an external file to JBrowse, add a trackMetadata section to the JBrowse configuration.
JBrowse currently supports track metadata that in Excel-compatible comma-separated-value (CSV) format, but additional track metadata backends are relatively easy to add. Write the JBrowse mailing list if you have a strong need to use another format.
| Option | Value |
|---|---|
trackMetadata.sources |
Array of source definitions, each of which takes the form { type: 'csv', url: '/path/to/file' }. The url is interpreted as relative to the url of the page containing JBrowse (index.html in default installations). Source definitions can also contain a class to explicitly specify the JavaScript backend used to handle this source. |
trackMetadata.indexFacets |
Optional array of facet names that should be the only ones made searchable. This can be used improve the speed and memory footprint of JBrowse on the client by not indexing unused metadata facets. |
Hint: to convert JBrowse JSON files to a csv, try using jq https://stedolan.github.io/jq
Example:
cat trackList.json| jq -r '.tracks[] | [.label,.key] | @csv'
Will produce a CSV with the label and key of each track in your trackList.json
Another for thing you can do with jq is add config variables directly to your trackList, for example
cat trackList.json| jq -r '.tracks[].maxExportSpan=50000'
Configuration:
"trackMetadata": {
"indexFacets": [ "category","organism","target","technique","principal_investigator",
"factor","developmental-stage","strain","cell-line","tissue","compound",
"temperature"
],
"sources": [
{ "type": "csv", "url": "myTrackMetaData.csv" }
]
}
Note: use lower case values for the facet names / column names in the CSV. Use renameFacets to give them other names. See #Faceted_track_selector for details.
Track metadata CSV:
| label | technique | factor | target | principal_investigator | submission | category | type | Developmental-Stage |
|---|---|---|---|---|---|---|---|---|
| fly/White_INSULATORS_WIG/BEAF32 | ChIP-chip | BEAF-32 | Non TF Chromatin binding factor | White, K. | 21 | Other chromatin binding sites | data set | Embryos 0-12 hr |
| fly/White_INSULATORS_WIG/CP190 | ChIP-chip | CP190 | Non TF Chromatin binding factor | White, K. | 22 | Other chromatin binding sites | data set | Embryos 0-12 hr |
| fly/White_INSULATORS_WIG/GAF | ChIP-chip | GAF | Non TF Chromatin binding factor | White, K. | 23 | Other chromatin binding sites | data set | Embryos 0-12 hr |
| ... | ... | ... | ... | ... | ... | ... | ... | ... |
Note that the label for each track metadata row must correspond to the label in the track configuration for the track it describes.
Hint to set track key from metadata: if a track has no key set in its track configuration, JBrowse will look for a key in its track metadata, and use that if it is present.
trackMetadata.sources - array of metadata source objects
Each source can have
-url - a URL (relative to JBrowse root directory, or absolute URL) -type - can be CSV, JSON or something else. inferred from the filename of the URL if none specified -storeClass - can be any store class, defaults to 'dojox/data/CsvStore' for CSV type and 'dojox/data/JsonRestStore' for JSON type
Example:
"trackMetadata": {
"sources": [
{ "type": "csv", "url": "data/myTrackMetaData.csv" }
]
}
This would load data/myTrackMetaData.csv, e.g. from your data folder. Note trackMetadata blocks can be specified in the trackList.json or a global config file
Starting with version 1.4.0, JBrowse has an optional "faceted" track selector designed for sites with hundreds or even thousands of tracks in a single JBrowse instance. This track selector allows users to interactively search for the tracks they are interested in based on the metadata for each track.
An example of a faceted track selector in action with about 1,800 tracks can be seen here. This is an example installation containing a snapshot of modENCODE track metadata. Note that the track data and reference sequences in this example are not real (they are actually all just copies of the same volvox test track), this is just an example of the faceted track selector in action.
The Faceted track selector takes all sources of track metadata, aggregates them, and makes the tracks searchable using this metadata. By default, tracks only have a few default metadata facets that come from the track configuration itself. After initially turning on the faceted track selector, most users will want to add their own metadata for the tracks: see Defining Track Metadata below. To enable the faceted track selector in the JBrowse configuration, set trackSelector→type to Faceted.
There are some other configuration variables that can be used to customize the display of the track selector. Most users will want to set both of these variables to customize the columns and facets shown in the track selector.
| Option | Value |
|---|---|
trackSelector→displayColumns |
Array of which facets should be displayed as columns in the track list. Columns are displayed in the order given. If not provided, all facets will be displayed as columns, in lexical order. |
trackSelector→renameFacets |
Object containing "display names" for some or all of the facet names. For example, setting this to { "developmental-stage": "Dev. Stage" } would display "Dev. Stage" as the name of the developmental-stage facet. |
trackSelector→escapeHTMLInData |
Beginning in JBrowse 1.6.5, if this is set to true or 1 prevents HTML code that may be present in the track metadata from being rendered. Instead, the HTML code itself will be shown. |
trackSelector→selectableFacets |
Optional array of which facets should be displayed as facet selectors. The selectors for these appear in the order in which they are specified here. *Note: the names of the facets are required to be in all lower case for selectableFacets to work* |
trackSelector→initialSortColumn |
A column specifying how the faceted selector is initially sorted. This parameter should be the name used in the displayColumns array and not the "renamed" facet column name. |
"trackSelector": {
"type": "Faceted",
"displayColumns": [
"key",
"organism",
"technique",
"target",
"factor",
"developmental-stage",
"principal_investigator",
"submission"
],
"renameFacets": { "developmental-stage": "Conditions", "submission": "Submission ID" },
"selectableFacets": ["organism","technique","developmental-stage","factor"]
}
Starting with version 1.11.0, JBrowse uses a "Hierarchical" track selector by default that is designed to allow grouping of tracks and easy checkbox selection choices. The Hierarchical track selector is like the old Simple track selector (which was the default), except it pays attention to the “category” key in a track’s metadata, and if it is present, it organizes the tracks into nested, collapsible panes based on that. Also, instead of the drag-and-drop paradigm used by the Simple track selector, the Hierarchical track selector turns tracks on and off by just checking and unchecking the box next to each track. For more information, see 1
To assign categories and subcategories to your tracks, set category or metadata.category attributes on each configured tracks in your trackList.json. Starting in JBrowse 1.11.5, the category can also be read from a trackMetadata.csv category columns.
There are some other configuration variables that can be used to customize the display of the Hierarchical track selector.
| Option | Value |
|---|---|
trackSelector→sortHierarchical |
Can be true or false. If true, categories and tracks are sorted in alphabetical order. If false, tracks will be loaded specifically in the order that they are specified in the tracklist configuration files. Default:true. Added in JBrowse 1.11.5 |
trackSelector→collapsedCategories |
A comma separated list of categories from the trackList that will be collapsed initially. This helps when many tracks are loaded in the trackList but you want to collapse certain categories when the user first loads. If there are subcategories specified using slashes, don't use spaces around the slashes. Default:none. Added in JBrowse 1.11.5 |
JBrowse supports some other configuration variables that customize the overall behavior of the browser. Each of these variables goes in the top level of JSON configuration files (i.e. jbrowse_conf.json), or in a [general] section in textual configuration files (i.e. jbrowse.conf), or outside of the "tracks" section in a trackList.json or tracks.conf.
| Option | Description |
|---|---|
locationBoxLength |
The desired size, in characters of the location search box. If not set, the size of the location box is calculated to fit the largest location string that is likely to be produced, based on the length of the reference sequences and the length of their names. Added in JBrowse 1.7.0. |
css |
Used to add additional CSS code to the browser at runtime. Can be an array of either strings containing CSS statements, or URLs for CSS stylesheets to load (as json:{url: "/path/to/my.css"}). CSS can of course also be added outside of JBrowse, at the level of the HTML page where JBrowse runs. Added in JBrowse 1.6.2. |
theme |
Allows changing the graphical theme from the default Dijit "tundra" theme. Added in JBrowse 1.7.0. Intended primarily for use by plugin implementors or developers integrating JBrowse into a larger system. |
defaultTracks |
Comma-separated list of track *labels* to show on initial load if a user has never visited this browser before. Example: "snps,genes,alignments". |
forceTracks |
Comma-separated list of track *labels* to show on initial load, regardless of the user's saved settings, but which is overridden by the URL list. Example: "snps,genes,alignments". |
defaultLocation |
Initial location to be shown for users who have never visited the browser before. Example: "ctgA:1234..5678". |
view→trackPadding |
Spacing between tracks in the genome view, in pixels. Default 20. Added in JBrowse 1.10.10. |
include |
Imports one or more other configuration files and merges their data with the file in which this appears. Can be either a string URL of a single file, or an array of string URLs. |
initialHighlight |
If set, the given region will be highlighted when the browser loads. Example: "ctgA:1234..5678". |
highResoutionMode |
Enable high-resolution canvas rendering. Can be "auto", "disabled", or a numeric specifying a scaling factor. Default "disabled". Added in JBrowse 1.11.4. |
documentDomain |
Set the document.domain property which can assist when JBrowse is in embedded mode and needs to set the same-origin policy. Added in JBrowse 1.11.3. |
shareLink |
Enable or disable the share link button. Default: true. Can be 0 or false. |
shareURL |
A customized shareURL for the share link button. This can be helpful for an iframe embedding to have the share link point to the page embedding the iframe. A customized example could be "function(browser){ return 'http://myinstance.com/?loc='+browser.view.visibleRegionLocString(); }" |
aboutThisBrowser→title |
Title for the "About" popup. |
aboutThisBrowser→description |
Custom content for the "About" popup. |
maxRecentTracks |
Max length of the recent tracks menu. Default: 10 |
show_tracklist |
Optionally turn off tracklist. &tracklist=0 in the URL also works. Default: true |
show_nav |
Optionally turn off navigation bar. &nav=0 in the URL also works. Default: true |
show_tracklabels |
Optionally turn off the track labels. Works when HideTrackLabels plugin is enables. Set &tracklabels=0 in the URL also works. Default: true |
show_fullviewlink |
Optionally turn off the "Full-view" link in embedded mode. Set &fullviewlink=0 in the URL. Default: true |
show_overview |
Optionally turn off the overview bar. &overview=0 in the URL also works. Default: true |
show_menu |
Optionally turn off the menu bar. &menu=0 in the URL also works. Added in 1.11.6. Default: true. |
containerID |
Set a prefix on cookies and identify the ID of the div holding JBrowse. Default: GenomeBrowser. |
cookieSizeLimit |
Default: 1200 |
updateBrowserURL |
Enable updating the URL with current view. If you encounter security exceptions from cross domain updating the URL, try disabling. Default: true |
view→maxPxPerBp |
Sets the maximum zoom level in pixels per basepair units. Default: 20 |
quickHelp→content |
Sets the content of the help dialog. |
quickHelp→href |
Sets the content of the help dialog using XHR to retrieve content. |
quickHelp→title |
Sets the title of the help dialog. Note: other dijit Dialog parameter can also be passed to quickHelp, these are just the basics. |
exactReferenceSequenceNames |
Disables the regularizeReferenceSequence routine. Default: false |
refSeqs |
A custom URL for your refSeqs.json file (or a webservice returning something in refSeqs.json format). Example: "http://mysite/refseqs.json". Can also be a URL for a FASTA index file in 1.12.0+. |
alwaysOnTracks |
Comma-separated list of track *labels* to always show when the browser is opened. Example: "snps,genes,alignments". |
classicMenu |
Boolean to use classic menu style (File) menu. Added in 1.12.0 |
hideGenomeOptions |
Boolean - Hide the options for opening a sequence file. Added in 1.12.0 |
noPluginsForAboutDialog |
Boolean - Hide the plugins from the about box. Added in 1.12.3 |
trackLabels |
Set "trackLabels": "no-block" to enable the trackLabels to be out of the way of the features. Added in 1.12.5 |
General configuration options that can be added to a track
| Option | Description |
|---|---|
style->trackLabelCss |
Add arbitrary CSS to the track label |
style->histCss |
Add arbitrary CSS to the histogram. Used for HTMLFeatures histograms only |
style->featureCss |
Add arbitrary CSS to the features. Used for HTMLFeatures features only |
storeTimeout |
Add timeout for calculating the feature track statistics. Mostly applicable to VCF and BAM file for when you get the error "Too many BAM features. BAM chunk size M bytes exceeds chunkSizeLimit of N" |
Beginning in version 1.9.0, JBrowse supports an optional dropdown dataset selector that appears in the menu bar in the upper left corner of the JBrowse display. The selector displays a list of dataset names based on the datasets key in the JBrowse configuration. When the selector is changed, a page reload is triggered, with the browser navigating to the URL associated with the selected dataset.
To set which value of the dataset selector is currently selected, the configuration variable dataset_id should be set to the name of the corresponding entry in the datasets list.
The selector is only shown if both datasets and dataset_id are set in the configuration.
In a global config, like jbrowse.conf, you can add the "list of datasets"
in jbrowse.conf:
[datasets.volvox]
url = ?data=sample_data/json/volvox
name = Volvox Example
[datasets.modencode]
url = ?data=sample_data/json/modencode
name = MODEncode Example
[datasets.yeast]
url = ?data=sample_data/json/yeast
name = Yeast Example
Then add the "dataset_id" to the tracks.conf file for your individual data directories
in sample_data/json/volvox/tracks.conf:
[general]
dataset_id = volvox
in sample_data/json/modencode/tracks.conf:
[general]
dataset_id = modencode
in sample_data/json/yeast/tracks.conf:
[general]
dataset_id = yeast
You can see that the dataset_id corresponds to whatever is inside the declaration, e.g. when it says [datasets.yeast], yeast is the ID.
Note that it is also possible to put the "list of datasets" in a tracks.conf file instead of the jbrowse.conf file if you prefer to store all your configs in track specific directories. Then you just paste the same "list of datasets" in all your dataset directories.
For example, having this code in tracks.conf
[general]
dataset_id = fish1
[datasets.fish1]
name = Fish genome 1.0
[datasets.fish2]
name = Fish genome 2.0
Will populate the dataset selector with Fish 1 and 2 and display the name of Fish 1 in the genome area. This is just in contrast to putting the datasets configuration in jbrowse.conf and the dataset_id in the individual data directories (e.g. all the datasets configs are copied to each data directory)
By default, JBrowse instances report usage statistics to the JBrowse developers. This data is very important to the JBrowse project, since it is used to make the case to grant agencies for continuing to fund JBrowse development. No research data is transmitted, the data collected is limited to standard Google Analytics, along with a count of how many tracks the JBrowse instance has, how many reference sequences are present, their average length, and what types of tracks (wiggle, feature, etc) are present. Users can disable usage statistics by setting suppressUsageStatistics: true in the JBrowse configuration.
More than 40 third-party plugins are available that extend or change JBrowse's functionality. For a list of them, see https://gmod.github.io/jbrowse-registry/.
Plugins can be manually copied into the JBrowse `plugins` directory. For example, if you had a plugin called MyAwesomePlugin, you would install it by copying it into the JBrowse-1.x.x/plugins/MyAwesomePlugin directory.
Note: Beginning in JBrowse 1.13.0, you need to use the `*-dev.zip` build of JBrowse if you use any plugins beyond the default ones, and any time you add or change a plugin, you must re-run setup.sh to build the plugin's code into JBrowse.
Also note that plugins that are installed are not necessarily activated for every configuration in a JBrowse installation, they are just available.
To activate a plugin, add a plugins configuration variable in your jbrowse_conf.json file in the top-level JBrowse directory, and add an entry telling JBrowse the names of the plugins to load.
Example:
// array of strings (will look in JBrowse-1.x.x/plugins/MyAwesomePlugin)
"plugins": [ 'MyAwesomePlugin' ]
or in the text .conf format:
plugins =
+ MyAwesomePlugin
+ PubAnnotation
The JBrowse formatting tools biodb-to-json.pl and prepare-refseq.pl can extract data from existing databases that are supported by BioPerl Bio::DB::* adapters, such as GBrowse databases created by bp_seqfeature_load.pl, or Chado databases. Both tools accepts a configuration file in JSON format that contains the details about how to connect to a given database, and which data to extract from it, and which JBrowse feature tracks to create to display the data.
For example, to extract data from a Chado schema in PostgreSQL, one might start with a configuration like:
{
"description": "D. melanogaster (release 5.37)",
"db_adaptor": "Bio::DB::Das::Chado",
"db_args": { "-dsn": "dbi:Pg:dbname=fruitfly;host=localhost;port=5432",
"-user": "yourusername",
"-pass": "yourpassword"
},
...
}
In the database source name (dsn) argument, 'dbi:Pg' indicates that you are using PostgreSQL, and the dbname, host, and port were specified when the database was created with PostgreSQL's createdb command. The user and pass arguments were specified when the PostgreSQL user account was created with the createuser command. Collectively, these arguments identify the database and give the Bio::DB::Das::Chado object access to it. Other adaptors (Bio::DB::SeqFeature::Store, Bio:DB::GFF, etc.) will require similar information.
Here is a sample configuration file, usable with biodb-to-json.pl and prepare-refseqs.pl, with each line explained. Note that, in order for this config file to work, it would be necessary to remove the grey comments (since JSON does not support them). Also, notice that the config file is divided into two parts, a header section that contains information about the database, and a body section that contains information about the feature tracks.
{
//This is the header. It contains information about the database.
// description: a brief textual description of the data source.
"description": "D. melanogaster (release 5.37)",
//db_adaptor: a perl module with methods for opening databases and extracting
//information. This will normally be either Bio::DB::SeqFeature::Store,
//Bio::DB::Das::Chado, or Bio::DB::GFF, but it can also be the name of any
//other perl module that implements the Bio::SeqFeatureI interface.
"db_adaptor": "Bio::DB::SeqFeature::Store",
//db_args: arguments required to produce an instance of the db_adaptor. The
//required arguments can be found by searching for the db_adaptor on the CPAN
//website.
"db_args": {
//adaptor: With Bio::DB::SeqFeature::Store, a value of "memory"
//for the adaptor indicates that the data is stored somewhere in
//the file system. Alternatively, it might have been stored in a
//database such as MySQL or BerkeleyDB.
"-adaptor": "memory",
//dir: given the "memory" argument for the adaptor, this is the
//file system path to the location in memory where the data is
//stored. Data will automatically be extracted from any *.gff
//or *.gff3 files in this directory.
"-dir": "/Users/stephen/Downloads/dmel_r5.37"
},
//This is the body. It contains information about the feature tracks.
//TRACK DEFAULTS: The default options for every track.
"TRACK DEFAULTS": {
//class: same as 'cssClass' in flatfile-to-json.pl.
"class": "feature"
},
//tracks: information about each individual track.
"tracks": [
//Information about the first track.
{
//track: same as 'tracklabel' in flatfile-to-json.pl.
"track": "gene",
//key: same meaning as in flatfile-to-json.pl.
"key": "Gene Span",
//feature: an array of the feature types that will be used for the track.
//Similar to 'type' in flatfile-to-json.pl.
"feature": ["gene"],
"class": "feature2",
//urlTemplate: same meaning as in flatfile-to-json.pl. Note how
//urlTemplate is being used with a variable called "feature_id" defined
//in extraData. In this way, different features in the same track can
//be linked to different pages on FlyBase.
"urlTemplate": "http://flybase.org/cgi-bin/fbidq.html?{feature_id}",
//extraData: same as in flatfile-to-json.pl.
"extraData": {"feature_id": "sub {shift->attributes(\"load_id\");}"}
},
//Information about the second track.
{
"track": "mRNA",
"feature": ["mRNA"],
//subfeatures: similar to 'getSubs' in flatfile-to-json.pl.
"subfeatures": true,
"key": "mRNA",
"class": "transcript",
//subfeature_classes: same as 'subfeatureClasses' in flatfile-to-json.pl.
"subfeature_classes": {
"CDS": "transcript-CDS",
"five_prime_UTR": "transcript-five_prime_UTR",
"three_prime_UTR": "transcript-three_prime_UTR"
},
//arrowheadClass: same meaning as in flatfile-to-json.pl.
"arrowheadClass": "transcript-arrowhead",
//clientConfig: same meaning as in flatfile-to-json.pl.
"clientConfig": {
"histScale":5
},
"urlTemplate": "http://flybase.org/cgi-bin/fbidq.html?{feature_id}",
"extraData": {"feature_id": "sub {shift->attributes(\"load_id\");}"}
}
]
}Adding a loading page can help if your JBrowse instance is loading a large amount of data over a slow connection. This page will be displayed on browsing to the page, up until the data is all loaded in the backend.
First, modify your index.html's main GenomeBrowser element, adding a div inside GenomeBrowser with a custom ID such as loading_screen
<div id="GenomeBrowser" style="height: 100%; width: 100%; padding: 0; border: 0;">
<div id="loading_screen" style="padding: 50px;">
<h1>Loading data</h1>
</div>
</div>
Next, in the same file, you'll need to edit the javascript to add a callback. After the line
JBrowse = new Browser( config );
You'll need to add the line
JBrowse.afterMilestone('loadRefSeqs', function() { dojo.destroy(dojo.byId('loading_screen')); });
The afterMilestone function registers a callback to be called once all of the background data has been loaded. Make sure the id mentioned in the javascript statement matches that of your loading div.
The previous section covered the afterMilestone function for waiting on certain tasks to be finished during initialization
Other milestones include:
- initPlugins - plugins loaded. note: plugins can be loaded before everything else is finished, so plugins may use these milestone functions in their own code
- loadUserCSS - user CSS loaded
- loadRefSeqs - reference sequence json or data is loaded
- loadNames - names store loaded
- initView - view initialized
- loadConfig - configurations loaded
- initTrackMetadata - track metadata loaded
- createTrack - when track list finished initializing
- completely initialized - all milestones passed. note the space in the milestone name
Files do not have to be in the default location. They can be in different locations, such as a mounted filesystem, using apache.
<VirtualHost #:443>
Alias /genomes /data/share/web_public/jbrowse_ucschg19/genomes
Alias /ucschg19 /data/share/web_public/jbrowse_ucschg19/reference_data
</VirtualHost>
and then your trackList.json
{
"tracks" : [
{
"style" : {
"className" : "generic_parent",
"featureCss" : "background-color: #1400fa; height: 6px;",
"histCss" : "background-color: #1400fa;"
},
"hooks" : {
"modify" : "function(track, feat, elem) {\n var fType = feat.get(\"Type\");\n if (fType) {\n elem.className = \"basic\";\n switch (fType) {\n case \"CDS\":\n case \"thick\":\n elem.style.height = \"10px\";\n elem.style.marginTop = \"-3px\";\n break;\n case \"UTR\":\n case \"thin\":\n elem.style.height = \"6px\";\n elem.style.marginTop = \"-1px\";\n break;\n }\n elem.style.backgroundColor = \"#1400fa\";\n }\n}\n"
},
"key" : "ucscGenePfam",
"urlTemplate" : "/ucschg19/tracks/ucscGenePfam/{refseq}/trackData.json",
"compress" : 0,
"label" : "ucscGenePfam",
"type" : "FeatureTrack",
},
{
"label" : "NAME_OF_STUDY_genomes",
"key" : "Name_of_study Genomes",
"storeClass" : "JBrowse/Store/SeqFeature/VCFTabix",
"urlTemplate" : "/genomes/NAME_OF_STUDY_genomes/{refseq}/NAME_OF_STUDY_{refseq}.vcf.gz",
"tbiUrlTemplate": "/genomes/NAME_OF_STUDY_genomes/{refseq}/NAME_OF_STUDY_{refseq}.vcf.gz.tbi",
"type" : "JBrowse/View/Track/HTMLVariants",
"noExport" : "true",
},
Classes modeling individual sequence features conform to the Feature API (exemplified by and documented in the source for the class JBrowse/Model/SimpleFeature) by providing accessor methods for various feature attributes (start and endpoint, ID field, tags, score, parent and child relationships for modeling super- and sub-features) some of which are mandatory for the various different types of track. Specifically: “start” and “end” attributes are always required (representing 1-based closed-interval coordinates), a unique “id” attribute is required by non-quantitative tracks (CanvasFeatures, HTMLFeatures, Alignments*, etc) and a “score” attribute is used by quantitative tracks (Wiggle/*) to represent the score for the interval spanned by the feature.
By contrast, classes modeling sources of sequences and sequence features generally inherit from JBrowse/Store/SeqFeature and implement the Feature Store API including methods getGlobalStats (for global statistics about the features in the store), getRegionStats (for statistics about a particular interval), getFeatures (to query the store for features) and getReferenceSequence (to query the store for sequence data).
Typically, different Feature Stores will provide their own custom implementations of the Feature API.
Three track lists are currently implemented: JBrowse/View/TrackList/Simple, JBrowse/View/TrackList/Hierarchical, and JBrowse/View/TrackList/Faceted.
The Hierarchical and Faceted track lists use the track metadata store (JBrowse/Store/TrackMetaData), which is instantiated and kept by the Browser object, for access to track metadata (defined as specified in the “Data Sources” section). In particular, the category used by the hierarchical track selector is a piece of track metadata called “category”.
The JBrowse/GenomeView class manages the main genome view, where the tracks are displayed (see Figure 1). It manages showing and hiding tracks, reordering them, scrolling and zooming, and so forth.
Track objects query data from a feature store, and draw it in the GenomeView pane. The implementations are in JBrowse/View/Track/ for which the base class is JBrowse/View/Track/BlockBased.
CanvasFeatures tracks use various glyph subclasses, locatable in the JBrowse/View/FeatureGlyph/ directory and inheriting from the base class JBrowse/View/FeatureGlyph.
Users can extend JBrowse's functionality to with their own JavaScript code using the JBrowse plugin system. For an overview of plugins and their structure, see Writing JBrowse Plugins.
To use JBrowse with an existing set of web services, users will want to implement a JBrowse Store module in JavaScript that can fetch data from them and convert it into the internal JavaScript object representations that JBrowse expects. In general terms, the steps to follow to do this would be:
- Create a new plugin using
bin/new-plugin.plor manually. - Enable the plugin by adding its name to
pluginsin the JBrowse configuration (in jbrowse_conf.json, trackList.json, in the constructor arguments in index.html, or elsewhere). - Create a new data store class in the plugin's JS directory that inherits from JBrowse/Store/SeqFeature and overrides its methods.
In plugins/MyPlugin/js/Store/SeqFeature/FooBaseWebServices.js, usable in store or track configurations as MyPlugin/Store/SeqFeature/FooBaseWebServices.
Note that the most basic class could simply have a "getFeatures" function that grabs the feature data.
/**
* Example store class that uses Dojo's XHR libraries to fetch data
* from backend web services. In the case of feature data, converts
* the data into JBrowse SimpleFeature objects (see
* JBrowse/Model/SimpleFeature.js) but any objects that support the
* same methods as SimpleFeature are fine.
*/
define([
'dojo/_base/declare',
'dojo/_base/array',
'dojo/request/xhr',
'JBrowse/Store/SeqFeature',
'JBrowse/Model/SimpleFeature'
],
function( declare, array, xhr, SeqFeatureStore, SimpleFeature ) {
return declare( SeqFeatureStore, {
constructor: function( args ) {
// perform any steps to initialize your new store.
},
getFeatures: function( query, featureCallback, finishCallback, errorCallback ) {
var thisB = this;
xhr.get( this.config.baseUrl+'my/features/webservice/url',
{ handleAs: 'json', query: query }
).then(
function( featuredata ) {
// transform the feature data into feature
// objects and call featureCallback for each
// one. for example, the default REST
// store does something like:
array.forEach( featuredata || [],
function( featureKeyValue ) {
var feature = new SimpleFeature({
data: featureKeyValue
});
featureCallback( feature );
});
// call the endCallback when all the features
// have been processed
finishCallback();
},
errorCallback
);
}
});
});
Note: other feature stores can be "derived from" or extended in different ways. The FeatureCoverage store class is a good example of a store class that uses the BAM store, but instead overrides the functionality to calculate coverage.
Beginning in version 1.9.0, JBrowse ships with a REST data store adapter (JBrowse/Store/SeqFeature/REST) that can provide feature, sequence, and quantitative data for display by any of JBrowse's track types. To use it, a developer can implement server-side web services that satisfy the REST API it expects.
JBrowse version 1.11.0 added a REST adaptor that can look up names and name prefixes (for type-ahead completion) from REST endpoints as well (see JBrowse REST Names API below).
The JBrowse REST feature store requires the following server resources.
Required. Returns a JSON object containing global statistics about the features served by this store.
Example:
{
"featureDensity": 0.02,
"featureCount": 234235,
"scoreMin": 87,
"scoreMax": 87,
"scoreMean": 42,
"scoreStdDev": 2.1
}
None of the attributes in the example above are required to be present. However, if the store is primarily providing positional data (such as genes), it is recommended to provide at least featureDensity (average number of features per basepair), since JBrowse uses this metric to make many decisions about how to display features. For stores that primarily provide quantitative data, it is recommended to also provide score statistics.
Optional, but recommended. Get statistics for a particular region. Returns the same format as stats/global above, but with statistics that apply only to the region specified.
The refseq name URL component, and the start and end query parameters specify the region of interest. Statistics should be calculated for all features that overlap the region in question. start and end are in interbase coordinates.
NOTE: If this is not implemented, the statistics will be calculated as needed by actually fetching feature data for the region in question. If your backend *does* implement region stats, set "region_stats": true in the track or store configuration to have JBrowse use them.
Optional, added in JBrowse 1.10.7. Get binned feature counts for a certain region, which are used only by HTMLFeatures tracks to draw density histograms at certain zoom levels. If your backend implements this endpoint, set "region_feature_densities": true in the track or store configuration to have JBrowse use it.
The refseq name URL component, and the start and end query parameters specify the region of interest. start and end are in interbase coordinates.
The basesPerBin is an integer which must be used to determine the number of bins - the endpoint may not choose its own bin size. max should be the maximum value for the density, the global maximum for the entire track.
Example returned JSON:
{
"bins": [ 51, 50, 58, 63, 57, 57, 65, 66, 63, 61,
56, 49, 50, 47, 39, 38, 54, 41, 50, 71,
61, 44, 64, 60, 42
],
"stats": {
"basesPerBin": 200,
"max": 88
}
}
Note that the stats.max attribute sets that Y-axis scale for the entire track, so should probably be set according to the global (or nearly global) max count for bins of that size.
Required. Fetch feature data (including quantitative data) for the specified region.
The refseq name URL component, and the start and end query parameters specify the region of interest. All features that overlap the region in question should be returned. start and end are in interbase coordinates. Also, track types that display features as boxes laid out on the genome (such as HTMLFeatures, CanvasFeatures, Alignments, and Alignments2 ) require all top-level features to have a globally-unique ID, which should be set as uniqueID in the JSON emitted by the service. uniqueID can be any string or number that is guaranteed to be unique among the features being emitted by this query. It is never shown to the user.
Example return JSON:
{
"features": [
/* minimal required data */
{ "start": 123, "end": 456 },
/* typical quantitative data */
{ "start": 123, "end": 456, "score": 42 },
/* Expected format of the single feature expected when the track is a sequence data track. */
{"seq": "gattacagattaca", "start": 0, "end": 14},
/* typical processed transcript with subfeatures */
{ "type": "mRNA", "start": 5975, "end": 9744, "score": 0.84, "strand": 1,
"name": "au9.g1002.t1", "uniqueID": "globallyUniqueString3",
"subfeatures": [
{ "type": "five_prime_UTR", "start": 5975, "end": 6109, "score": 0.98, "strand": 1 },
{ "type": "start_codon", "start": 6110, "end": 6112, "strand": 1, "phase": 0 },
{ "type": "CDS", "start": 6110, "end": 6148, "score": 1, "strand": 1, "phase": 0 },
{ "type": "CDS", "start": 6615, "end": 6683, "score": 1, "strand": 1, "phase": 0 },
{ "type": "CDS", "start": 6758, "end": 7040, "score": 1, "strand": 1, "phase": 0 },
{ "type": "CDS", "start": 7142, "end": 7319, "score": 1, "strand": 1, "phase": 2 },
{ "type": "CDS", "start": 7411, "end": 7687, "score": 1, "strand": 1, "phase": 1 },
{ "type": "CDS", "start": 7748, "end": 7850, "score": 1, "strand": 1, "phase": 0 },
{ "type": "CDS", "start": 7953, "end": 8098, "score": 1, "strand": 1, "phase": 2 },
{ "type": "CDS", "start": 8166, "end": 8320, "score": 1, "strand": 1, "phase": 0 },
{ "type": "CDS", "start": 8419, "end": 8614, "score": 1, "strand": 1, "phase": 1 },
{ "type": "CDS", "start": 8708, "end": 8811, "score": 1, "strand": 1, "phase": 0 },
{ "type": "CDS", "start": 8927, "end": 9239, "score": 1, "strand": 1, "phase": 1 },
{ "type": "CDS", "start": 9414, "end": 9494, "score": 1, "strand": 1, "phase": 0 },
{ "type": "stop_codon", "start": 9492, "end": 9494, "strand": 1, "phase": 0 },
{ "type": "three_prime_UTR", "start": 9495, "end": 9744, "score": 0.86, "strand": 1 }
]
}
]
}
Example configuration for an HTMLFeatures track showing features from a REST feature store with URLs based at http://my.site.com/rest/api/base, and also adding "organism=tyrannosaurus" in the query string of all HTTP requests.
{
"label": "my_rest_track",
"key": "REST Test Track",
"type": "JBrowse/View/Track/HTMLFeatures",
"storeClass": "JBrowse/Store/SeqFeature/REST",
"baseUrl": "http://my.site.com/rest/api/base",
"query": {
"organism": "tyrannosaurus"
}
}
{
"tracks": [
{
"label": "my_gene_track", /* Unique, machine readable name */
"key": "Genes", /* Descriptive, meat readable name */
"type": "JBrowse/View/Track/HTMLFeatures",
"storeClass": "JBrowse/Store/SeqFeature/REST",
"baseUrl": "http://my.site.com/rest/api/base",
"query": { /* Your arbitrary set of query parameters, always sent with every request */
"organism": "tyrannosaurus", "soType": "gene"
}
},
{
"label": "my_sequence_track", /* Unique, machine readable name */
"key": "DNA", /* Descriptive, meat readable name */
"type": "JBrowse/View/Track/Sequence",
"storeClass": "JBrowse/Store/SeqFeature/REST",
"baseUrl": "http://my.site.com/rest/api/base",
"query": { /* Your arbitrary set of query parameters, always sent with every request */
"organism": "tyrannosaurus", "sequence": true
}
}
]
}
This will be fetched from the url configured in the config.json file, such that if the config.json file specifies "?data=X/Y/Z" and is itself at "SCHEME://HOST:PORT/PATH", then JBrowse will request the url "SCHEME://HOST:PORT/PATH/X/Y/Z/seq/refSeqs.json".
[
{"name": "chr1", "start": 0, "end": 12345678},
{...}
]
Retrieved from "{BASE}/features/{seqid}".
This is the REST feature store data format, but it expects just a single feature, and that feature should have sequence.
{"features": [
{"seq": "gattacagattaca" "start": 0, "end": 14}
]
Starting in version 1.11.0, JBrowse can use REST web services for looking up features by name, and for type-ahead autocompletion.
Required. Returns JSON list of genomic locations with names that exactly match the given string.
The JSON format returned is the same as for startswith above.
Required. Returns JSON list of genomic locations that have names that start with the given string.
Example returned JSON:
[
{
"name" : "Apple1", // Name associated with the record. May be a secondary name of the object.
"location" : { // location information for the match
"ref" : "ctgA", // name of the reference sequence
"start" : 9999, // genomic start (interbase coords)
"end" : 11500, // genomic end (interbase coords)
"tracks" : [ // list of track labels that contain this object
"CDS"
],
"objectName" : "Apple1" // canonical/primary name of the object
}
},
...
]
Add something like the following to jbrowse.conf:
[names]
type = REST
url = /path/to/names/rest/service
JBrowse client events are implemented using the dojo/topic message bus from the Dojo library. Extensions can subscribe to particular events in order to be notified when certain UI changes happen (for example, highlighting a region generates an event, which can be latched onto with a callback that triggers a request for the server to BLAST that region against a database). In select cases, extensions can also publish events, as a way of forcing the UI into certain states or transitions (for example, events can be used in this way to force the browser to load a new track, in response to some other circumstance or notification).
When events are associated with tracks, the event names take the form /jbrowse/v1/{type}/tracks/{trackEvent}. The {type} is one of ‘v’, ‘c’, or ‘n’, corresponding to view events (requests from the user, handled by the browser); command events (which trigger actions in the UI, and are often generated by the browser in response to view events); or notification events (informing subscribers that something just happened). The {trackEvent} specifies the nature of the track event and is one of ‘new’, ‘delete’, ‘show’, ‘hide’, ‘pin’, ‘unpin’, ‘replace’, ‘redraw’, or ‘visibleChanged’.
When events are global, the event names take the form /jbrowse/v1/{type}/{globalEvent} where {type} is as above and {globalEvent} is one of ‘navigate’ or ‘globalHighlightChanged’.
The DebugEvents plugin logs events to the console, and can be used to monitor events as they are triggered by user interactions with the browser.
Note that you can also publish and subscribe to a JBrowse instance that is in an iframe. See http://gmod.org/wiki/JBrowse_FAQ#How_can_I_get_jbrowse_to_update_the_URL_of_a_parent_page_when_jbrowse_is_inside_of_an_iframe for an example
The JBrowse plugin system allows users to write their own JavaScript classes and AMD modules to extend JBrowse in nearly any way imaginable. Just a few things that can be done by plugins:
- Add new data store adapters to allow JBrowse to read data from existing web services.
- Add new track classes for custom track displays and behavior (WebApollo is one well-known plugin that does this).
- Add (or remove) options in the JBrowse dropdown menus.
- Add new types of track selectors.
Also see this FAQ entry http://gmod.org/wiki/JBrowse_FAQ#How_do_I_create_a_plugin.3F and http://gmod.org/wiki/JBrowse_FAQ#What_is_a_plugin_useful_for.3F
For an example plugin named MyPlugin, all of its files should be located in plugins/MyPlugin in the main JBrowse directory. There are no restrictions on what a plugin directory may contain.
A plugin is required to have a plugins/MyPlugin/js/main.js file, which is an AMD module that returns a JavaScript "class" that inherits from the JBrowse/Plugin class. JBrowse will create one instance of this class, which will persist during the entire time JBrowse is running. This class's constructor function is the entry point for your plugin's JavaScript code.
If a plugin contains custom CSS rules, it can optionally have a plugins/MyPlugin/css/main.css file as well, which JBrowse will load asynchronously. If your plugin code needs to know when the CSS is finished loading, the cssLoaded property of the plugin object contains a Dojo Deferred that is resolved when the CSS load is complete. Multiple CSS files can be loaded using @import statements in the main.css.
JBrowse also defines an AMD namespace that maps to your plugin's js directory, which your plugin code (and JBrowse itself) can use to load additional AMD modules and JavaScript classes. For example, our MyPlugin example could have a data-store adaptor in the file plugins/MyPlugin/js/Store/SeqFeature/FooBaseServices.js, which would be loaded with the module name "MyPlugin/Store/SeqFeature/FooBaseServices".
Here is an example plugin main.js, implemented using the Dojo declare class system.
define([
'dojo/_base/declare',
'JBrowse/Plugin'
],
function(
declare,
JBrowsePlugin
) {
return declare( JBrowsePlugin,
{
constructor: function( args ) {
var browser = this.browser;
/* do anything you need to initialize your plugin here */
}
});
});
plugins/MyPlugin/js
plugins/MyPlugin/js/main.js
plugins/MyPlugin/js/MyPlugin.profile.js
plugins/MyPlugin/css
plugins/MyPlugin/css/main.css
plugins/MyPlugin/img
plugins/MyPlugin/img/myimage.png
The bin/new-plugin.pl will initialize a directory structure for you, e.g. run
bin/new-plugin.pl MyPlugin
JBrowse 1.13.0 and higher can also use plugins that are distributed via npm. To distribute your plugin on npmjs.org, you just need to add a package.json file to the top-level directory of your plugin, and your plugin's npm package name must end in '-jbrowse-plugin'. Note that the plugin is named "my-jbrowse-plugin" on npm, and includes a "jbrowsePlugin"
Here is an example package.json file for MyPlugin:
{
"name": "my-jbrowse-plugin",
"version": "1.0.0",
"description": "JBrowse client plugin for adding amazing things",
"main": "js/main.js",
"author": "Josephina Example",
"license": "SEE LICENSE FILE IN LICENSE.md",
"dependencies": {
},
"devDependencies": {
},
"repository": {
"type": "git",
"url": "git+https://github.com/josephina/myplugin.git"
},
"keywords": [
"genome",
"jbrowse"
],
"bugs": {
"url": "https://github.com/josephina/myplugin/issues"
},
"homepage": "https://github.com/josephina/myplugin#readme",
"jbrowsePlugin": {
"name": "MyPlugin"
}
}
Most of the time, simply putting plain javascript files in the plugins directory is enough. The AMD module loader will locate your plugin without issues. But there are a couple of scenarios where creating a minified distribution of your plugin is helpful. A major scenario is that your plugin uses dojo modules that JBrowse does not itself use. If your plugin uses dojo modules that are not used by JBrowse, then your plugin will not be usable in the "minified" release version of JBrowse since the unused code checker would have removed those unused modules. Therefore, you can create a custom build of your plugin that packages your plugin's dojo dependencies itself.
To fix these problems, you can run the JBrowse makefile which will automatically run the build steps needed to package your plugin
Simply run this command in your jbrowse root directory (must be a full github clone of jbrowse):
make -f build/Makefile release-notest
Note that for this build step to complete, you will need a "build profile" e.g. plugins/MyPlugin/js/MyPlugin.profile.js. This file is automatically scaffolded by bin/new-plugin.pl and an example is also available in the RegexSequenceSearch plugin. As noted above, you'll need a github clone of jbrowse or at the very least the full dojo SDK in order to run this build cycle (because the src/dojo/dojo.js in the release versions doesn't contain the build SDK).
After creating your custom build, you can use it in release versions of JBrowse
One data store type that JBrowse uses is a lazily-loaded nested containment list (LazyNCLists), which is an efficient format for storing feature data in pre-generated static JSON files. A nested containment list is a tree data structure in which the nodes of the tree are intervals themselves features, and edges connecting features that lie `within the bounds of (but are not subfeatures of) another feature. It has some similarities to an R tree. For more on NClists, see the Alekseyenko paper.
This data format is currently used in JBrowse 1.3 for tracks of type FeatureTrack, and the code that actually reads this format is in SeqFeatureStore/NCList.js and ArrayRepr.js.
The LazyNCList format can be broken down into two distinct subformats: the LazyNCList itself, and the array-based JSON representation of the features themselves.
For speed and memory efficiency, NCList JSON represents features as arrays instead of objects. This is because the JSON representation is much more compact (saving a lot of disk space), and many browsers significantly optimize JavaScript Array objects over more general objects.
Each feature is represented as an array of the form [ class, data, data, ... ], where the class is an integer index into the store's classes array (more on that in the next section). Each of the elements in the classes array is an array representation that defines the meaning of each of the the elements in the feature array.
An array representation specification is encoded in JSON as (comments added):
{
"attributes": [ // array of attribute names for this representation
"AttributeNameForIndex1",
"AttributeNameForIndex2",
...
],
"isArrayAttr": { // list of which attributes are themselves arrays
"AttributeNameForIndexN": 1,
...
}
}
A JBrowse LazyNCList is a nested containment list tree structure stored as one JSON file that contains the root node of the tree, plus zero or more "lazy" JSON files that contain subtrees of the main tree. These subtree files are lazily fetched: that is, they are only fetched by JBrowse when they are needed to display a certain genomic region.
On disk, the files in an LazyNCList feature store look like this:
# stats, metadata, and nclist root node
data/tracks/<track_label>/<refseq_name>/trackData.json
# lazily-loaded nclist subtrees
data/tracks/<track_label>/<refseq_name>/lf-<chunk_number>.json
# precalculated feature densities
data/tracks/<track_label>/<refseq_name>/hist-<bin_size>.json
...`
Where the trackData.json file is formatted as (comments added):
{
"featureCount" : 4293, // total number of features in this store
"histograms" : { // information about precalculated feature-frequency histograms
"meta" : [
{ // description of each available bin-size for precalculated feature frequencies
"basesPerBin" : "100000",
"arrayParams" : {
"length" : 904,
"chunkSize" : 10000,
"urlTemplate" : "hist-100000-{Chunk}.json"
}
},
... // and so on for each bin size
],
"stats" : [
{ // stats about each precalculated set of binned feature frequencies
"basesPerBin" : "100000", // bin size in bp
"max" : 51, // max features per bin
"mean" : 4.93030973451327 // mean features per bin
},
...
]
},
"intervals" : {
"classes" : [ // classes: array representations used in this feature data (see ArrayRepr section above)
{
"isArrayAttr" : {
"Subfeatures" : 1
},
"attributes" : [
"Start",
"End",
"Strand",
"Source",
"Phase",
"Type",
"Id",
"Name",
"Subfeatures"
]
},
...
{ // the last arrayrepr class is the "lazyClass": fake features that point to other files
"isArrayAttr" : {
"Sublist" : 1
},
"attributes" : [
"Start",
"End",
"Chunk"
]
}
],
"nclist" : [
[
2, // arrayrepr class 2
12962, // "Start" minimum coord of features in this subtree
221730, // "End" maximum coord of features in this subtree
1 // "Chunk" (indicates this subtree is in lf-1.json)
],
[
2, // arrayrepr class 2
220579, // "Start" minimum coord of features in this subtree
454457, // "End" maximum coord of features in this subtree
2 // "Chunk" (indicates this subtree is in lf-2.json)
],
...
],
"lazyClass" : 2, // index of arrayrepr class that points to a subtree
"maxEnd" : 90303842, // maximum coordinate of features in this store
"urlTemplate" : "lf-{Chunk}.json", // format for lazily-fetched subtree files
"minStart" : 12962 // minimum coordinate of features in this store
},
"formatVersion" : 1
}
JBrowse can display tracks composed of precalculated image tiles, stretching the tile images horizontally when necessary. The JBrowse Volvox example data has a wiggle data track that is converted to image tiles using the included wig2png program, but any sort of image tiles can be displayed if they are laid out in this format.
The files for a tiled image track are structured by default like this:
data/tracks/<track_label>/<refseq_name>/trackData.json
data/tracks/<track_label>/<refseq_name>/<zoom_level_urlPrefix>/<index>.png
... (and so on, for many more PNG image files)
Where the PNG files are the image tiles themselves, and trackData.json contains metadata about the track in JSON format, including available zoom levels, the width and height of the image tiles, their base resolution (number of reference sequence base pairs per image tile), and statistics about the data (such as the global minimum and maximum of wiggle data).
The structure of the trackData.json file is:
{
"tileWidth": 2000, // width of all image tiles, in pixels
"stats" : { // any statistics about the data being represented
"global_min": 100,
"global_max": 899
},
"zoomLevels" : [ // array describing what resolution levels are available
{ // in the precalculated image tiles
"urlPrefix" : "1/",
"height" : 100,
"basesPerTile" : 2000
},
... (and so on, for zoom levels in order of decreasing resolution / increasing bases per tile )
]
}
To see a working example of this in action, see the contents of sample_data/json/volvox/tracks/volvox_microarray.wig/ctgA after the Volvox wiggle sample data has been formatted.
The code for working with this tiled image format in JBrowse 1.3 is in TiledImageStore/Fixed.js.
The trackList.json configuration format is limited when it comes to specifying callbacks, because functions can only be specified on a single line. However, you can create functions in an external .conf file that span multiple lines and include them in the trackList.json configuration easily. The functions should follow the guidelines specified in the .conf section here.
Example: say there is a complex coloring function, so it is stored in a file called functions.conf in the data directory
# functions.conf
customColor = function(feature) {
return feature.get("type")=="mRNA" ? "green" : "blue";
/* note the closing bracket should be spaced away from the leftmost column */
}
Then you can use this function in a particular track by referencing it with curly brackets, or "variable interpolation".
"style": {
"color":"{customColor}"
}
Make sure to also include your functions.conf in the "trackList.json" (e.g. anywhere outside the "tracks": [ ... ] section of trackList.json), add
"include": "functions.conf"
Note that include can also be an array of multiple files
"include": ["functions1.conf","functions2.conf"]
In the above example, the callback parameters exactly match, so the interpolated function can just be dropped in place. Alternatively, if the callback parameters don't match, you can store the interpolated function in a variable and adjust the callback parameters appropriately.
"style": {
"color": "function(feature) { var f={customColor}; return f(feature); }"
}
or shorthand
"style": {
"color": "function(feature) { return ({customColor})(feature); }"
}
See the general configuration section for details on the include command.
Using the command line tool phantomjs, you can produce a screenshot of JBrowse which includes high resolution canvas features. First get phantomJS and rasterize.js (http://phantomjs.org/screen-capture.html).
Then produce the screenshot using the syntax phantomjs rasterize.js <url> <output file> <dimensions> <zoom factor>
phantomjs rasterize.js 'http://your.jbrowse/?loc=ctgA:1..10000&tracks=DNA,Genes' jbrowse.png '4800px*2600px' 4
phantomjs rasterize.js 'http://your.jbrowse/?loc=ctgA:1..10000&tracks=DNA,Genes' jbrowse.pdf
Caveats
- High resolution canvas features requires setting something like highResolutionMode=4 (match the zoom factor essentially). Reason being is that devicePixelRatio detection isn't supported so highResolutionMode=auto doesn't work.
- A bugfix for phantomJS was added in 1.11.5 for release versions of JBrowse, so previous releases of JBrowse may get blank screenshots in phantomjs
- PDF output works in PhantomJS 2.0+
- The default browser HTML form elements do not scale well, including those on the hierarchical track panel, so taking the screenshot with &tracklist=0 will look better. As an example about how the HTML form elements appear, see this showing the effect of different zoom factor levels, but the problem being that the form elements remain tiny at larger zoom levels 2
- The default timeout for rasterize.js is small (200 ms) and you can increase this if you are getting a blank screenshot
- When you are exporting PDF, avoid setting dimensions (instead of 4800px*2600px like for png, pdf can accept something like 11in*8in, but don't use this!) and the zoomscale argument for PDF is unnecessary either (you can increase highResolutionMode in the jbrowse config though). Instead, modify the viewportSize inside rasterize.js to something "screen like" such as width: 1200, height: 750
You can also convert the PhantomJS PDF to SVG using Inkscape. Simply import the PDF generated by PhantomJS into Inkscape and use "save as..." which by default exports SVG. Alternatively, use the Inkscape command line
inkscape --without-gui --file=input.pdf --export-plain-svg=output.svg
On some platforms, you may need to specify the full file paths for input and output files
To begin editing the SVG can be a little daunting as the webpage is a complex object but first steps are probably to run "Object->Ungroup" in Inkscape, and then "Edit->Deselect" since ungrouping automatically selects everything. The you'll be able to select individual components and edit as needed.
Using pageres, which is a wrapper for PhantomJS, has some commands like --scale to enable easier zoomFactor scaling and -d to increase the timeout.
Install pageres-cli with "npm install -g pageres-cli" and install phantomjs as well. Then you can use arguments
Examples:
pageres -d 5 "http://jbrowse.org/code/JBrowse-1.11.6/?data=sample_data/json/volvox&tracklist=0" --filename=jbrowse_scale1 --scale=1
pageres -d 5 "http://jbrowse.org/code/JBrowse-1.11.6/?data=sample_data/json/volvox&tracklist=0" --filename=jbrowse_scale2 --scale=2
pageres -d 5 http://jbrowse.org/code/JBrowse-1.11.6/?data=sample_data/json/volvox&tracklist=0" --filename=jbrowse_scale4 --scale=4
These pageres commands using the --scale argument can upscale content to make high-res images automatically, but using highResolutionMode: 2 or similar is still recommended for canvasfeatures.
- See WormBase.org's blog post about this topic http://blog.wormbase.org/2016/02/10/creating-hi-res-screenshots-in-jbrowse/
- The service http://phantomjscloud.com can automate the this screenshot process from the cloud as well, for example by simply providing a jbrowse URL to their service. See https://phantomjscloud.com/api/browser/v2/a-demo-key-with-low-quota-per-ip-address/?request={url:%22https://jbrowse.org/code/JBrowse-1.11.6/?data=sample_data/json/volvox%26tracks=DNA%2CTranscript%2Cvolvox_microarray_bw_density%2Cvolvox_microarray_bw_xyplot%2Cvolvox-sorted-vcf%2Cvolvox-sorted_bam_coverage%2Cvolvox-sorted_bam%22,renderType:%22png%22,renderSettings:{zoomFactor:2,viewport:{width:3300,height:2000}}} as an example, this generates the screenshot from jbrowse.org on the fly
- Plugin for allowing easy screenshots based on phantomjscloud https://github.com/bhofmei/jbplugin-screenshot
Example:
{kind=link}