Visualize your assembly with IGV
IGV: Integrative Genomics Viewer.
We will use this software to evaluate our assemblies, among others to view how the reads map to the assembly.
We will also add tracks with genome annotation and gaps.
It is simply finding a/the matching locus/area of a read on a sequence. You could think of it at the location of where your read would hybridize to your genome if you could do this experiment
Usually few mismatches are allowed (think about the consequences).
Reads can be mapped as paired or single. If paired is used, then the matching regions are defined by the insert size and the length of each read
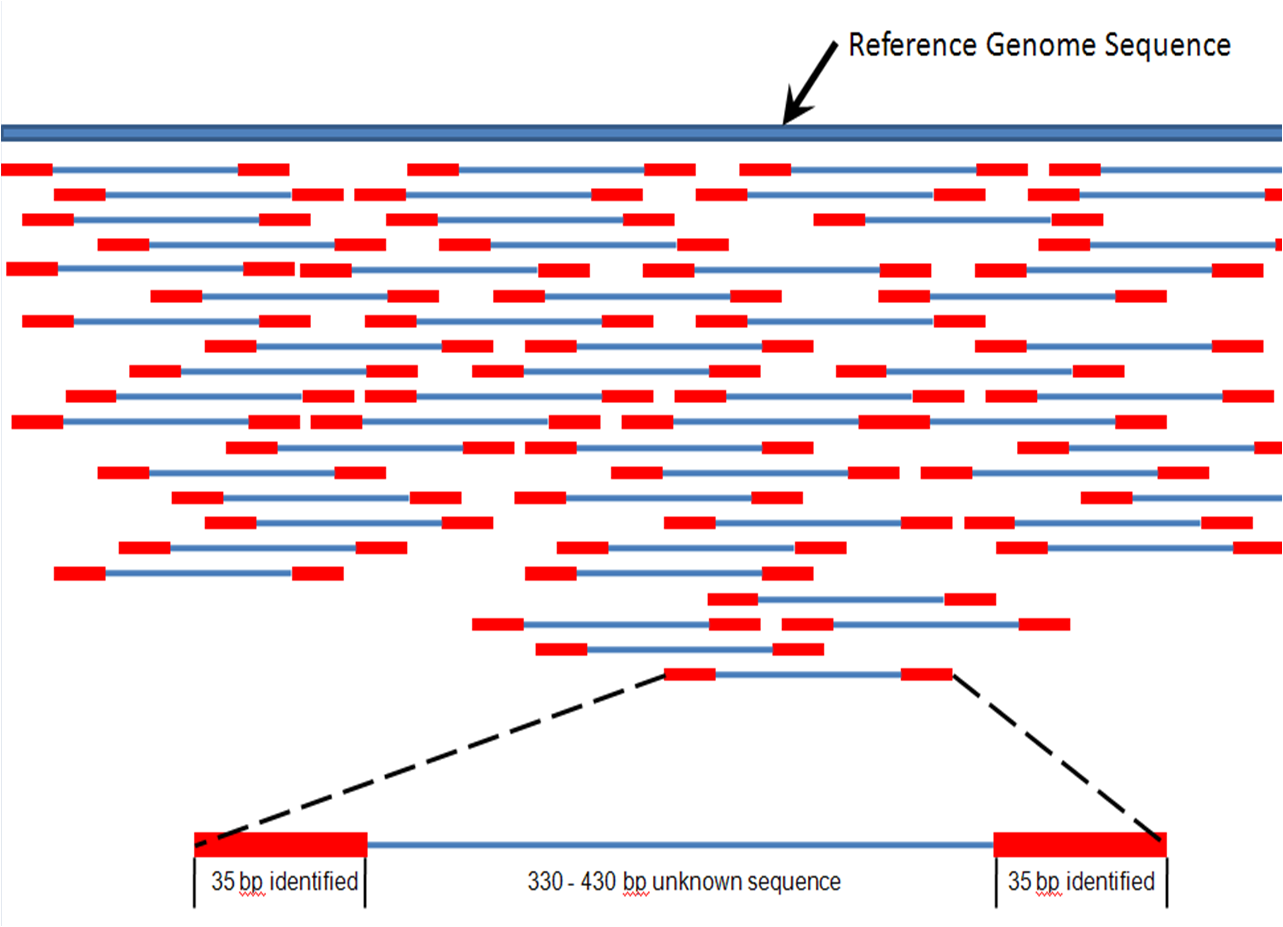
Read mapping techniques are used for several purposes. Some examples:
-
To evaluate the quality of an assembly (or to compare different methods used to assemble your reads). Read mapping can help identifying problematic areas. Indeed, statistics are necessary but might not be sufficient to evaluate the quality of your assembly.
We want to look at:
- the coverage regularity (ex: some repeated regions might have increased coverage)
- the coverage at the beginning and end of scaffolds - is it good enough?
- are they positions where pileup of reads show polymorphism?
- ...
-
To identify SNPs: some methods use reads mapping against a reference genome to identify and type SNPs
-
Assembly polishing software such as
pilonandreapruse mapped reads to identify potential improvement in assemblies. It is good to visualize what information they are using.Softwares can use variation in coverage, wrong read pairs orientation, discrepancy between expected insert size and actual insert size obtained from read mapping (ie. longer than expected) to improve assembly.
We use bwa mem option from bwa tools software.
We have assembled reads and annotated using Bifrost pipeline. The data for today's practical are found in /projects/nn9304k/bioinf_course/Mapping_training
You will use the trimmed-filtered reads in: /projects/nn9305k/bioinf_course/Mapping_training/bifrost_run/bbduk_trimmed and the assembly: /projects/nn9305k/bioinf_course/Mapping_training/bifrost_run/prokka/MiSeq_Ecoli_MG1655_50x.fna
We have to use the assembly output from Prokkato be able to visualize the assembly with IGV. (Scaffold names between assembly and annotations have to be consistent for the visualization).
Note that: Prokka transforms scaffold names from the assembly used as input. Annotation and assembly with annotation-matching scaffold names are provided as output files.
Practical:
In your project-home directory make a directory for today's work
and a folder called mapping where you will copy the input files
mkdir mapping
cd mapping
cp /projects/nn9305k/bioinf_course/Mapping_training/bifrost_run/bbduk_trimmed/*.fq.gz .
cp /projects/nn9305k/bioinf_course/Mapping_training/bifrost_run/prokka/MiSeq_Ecoli_MG1655_50x.fna .
# Also copy the annotation file .gff that we will need it later on
cp /projects/nn9305k/bioinf_course/Mapping_training/bifrost_run/prokka/MiSeq_Ecoli_MG1655_50x.gff .
# look at the file content (one reads-file, the assembly file):
head MiSeq_Ecoli_MG1655_50x.fna
gunzip -cd MiSeq_Ecoli_MG1655_50x_S_concat_stripped_trimmed.fq.gz | head
We will use the software included in the Bifrost pipeline. This is available as a conda environment, called bifrost.
qlogin --account=nn9305k --time=00:30:00 --ntasks=4 --mem-per-cpu=4G
source activate bifrost
# 1) We need to index the reference: we are in this case using the assembly as reference
bwa index <reference.fna>
# 2) We map the reads (attribute the position of the reads according to the index) as PE
bwa mem -t 4 <reference.fna> <in1:read1.fq.gz> <in2:read2.fq.gz> \
| samtools sort -o <out:PE_mapped_sorted.bam> -
# NB: We `sort` directly the mapping by index position
#`-` means that the output of the pipe is used as input in samtools
# For unpaired reads (called S here)
bwa mem -t 4 <reference.fna> <in:S_reads.fq.gz> \
| samtools sort -o <out:S_mapped_sorted.bam> -
# 3) We need to merge those two files as one
samtools merge <out:all_merged.bam> <in1:S_mapped_sorted.bam> <in2:PE_mapped_sorted.bam>
# 4) To be sure reads are still sorted: we resort
samtools sort -o <out:final_all_merged.bam> <in:all_merged.bam>OPTIONAL
Some software like Pilon need and updated index of mapped reads to run
(eg. after merging bam files). If necessary do:
samtools index <final_all_merged.bam>
1.4 The sam/bam file format
You can also have a look at Samtools article and at Samtools manual
.bam files are in a compressed binary format. We need to transform the .bam (to a .sam file) to be able to see how mapped-reads are represented in the file.
To decompress: chose f.eks. PE_mapped_sorted.bam that we did in the first step:
samtools view -h -o <out.sam> <in.bam>
Look at your .sam file with:
less <filename.sam>
1.5 looking at how the reads maps against the reference with Samtools tview module
# look at the reads pileup
samtools tview <final_all_merged.bam> --reference <assembly>
# -p <position> if you want to see a specific positiontype ? to view the navigation help while samtools is running
At your pc:
conda create -n <envname> -c bioconda igv
conda activate <IGV:envname>From the folder you want to work in transfer the following files from Abel to your pc:
- the assembly and annotation files
- the .bam file
scp <user_name>:<your_mapping_folder_assembly_AND_annotation_files> .We use a little python script from sequencetools repository to insert gap locations into a file and load it as a track in IGV. This will allow to easily locate the different scaffolds and potentially problematic regions. This is how we generate the file:
# On Abel: use biopython installed in conda bifrost to generate the .bed file
source activate bifrost
python /work/projects/nn9305k/vi_src/diverse/scaffoldgap2bed_py3.py -i <assembly.fna> > <gap_file>.bed
# From the folder you want to work in, transfer the .bed file from Abel to your pc
scp <user_name>:<your_mapping_folder_bed_file> .In Bifrost we annotated the assembly with Prokka (using annotations derived from a reference genome) .gff file. It contains the annotated gene locus tags that we use for visualization with IGV
2.4 Loading files in IGV

-
Create a
genome filethis allows associating tracks to the assembly :Genomes > create.genome file. Use the menu to select your assembly file.fastaand the annotation-gene file:.gff -
Load your
mapped readsand thegap fileusing:file > load from file -
To be able to easily re-open (without re-importing everything you can do):
file > save session
Now you are ready to navigate and explore your assembly.
Try to find a gap.
NB: To zoom while staying centered on the gap: click above menu (position within the scaffold - at the gap - top track) then click with the mouse at the gap position on the gap track (until appropriate zoom is obtained).
You can look here for Options and interpretation, and here: PE orientations.
Have a look at:
- coverage
- gaps positions
- some strange scaffolds?
- PE orientations: in detail how the reads map to your assembly (you will need to zoom a lot)
- are some PE reads miss-oriented? reported as having abnormal insert sizes?
You can look here at the Uio course for more details or if you want to do things slightly differently. We reuse some of their scripts.