a toolbox to analyze sequences, structures and simulations of RNA (and way more!)
Look for other our projects at https://github.com/RNA-Puzzles.



rna-tools is a core library and a set of programs to run various Python functions related to work, initially, with PDB files of RNA structures, but right now this is a huge toolbox of tools to process various types of RNA data.
That is why in 2019, after publishing our U6 Molecular Cell paper I decided to rename the package to rna-tools. Simply, various tools to work with RNA data: sequences, alignments, structures, trajectories, RNA-seq data. If you want access the old version see the branch.
The software is used by me in my servers NPDock (RNA/DNA-protein docking method, http://genesilico.pl/NPDock/) and SimRNAweb (RNA 3D structure prediction method, http://iimcb.genesilico.pl/SimRNAweb/) and mqapRNA (RNA 3D quality control, http://iimcb.genesilico.pl/mqapRNA/) and other projects EvoClustRNA and RNA-Puzzles-Normalized-submissions.
What is fun here?
rna-tools (formerly rna-pdb-tools) is a packages of shell utils that are using the common core library. You can also access functions of the library from your scripts.
A command-line tools:
$ rna_pdb_toolsx.py --is-pdb input/1I9V_A.pdb
True
$ rna_pdb_toolsx.py --is-pdb input/image.png
Falseor from a script:
>>> from rna_tools_lib import *
>>> s = RNAStructure('input/1I9V_A.pdb')
>>> s.is_pdb()
Trueor from a Jupyter Notebook:
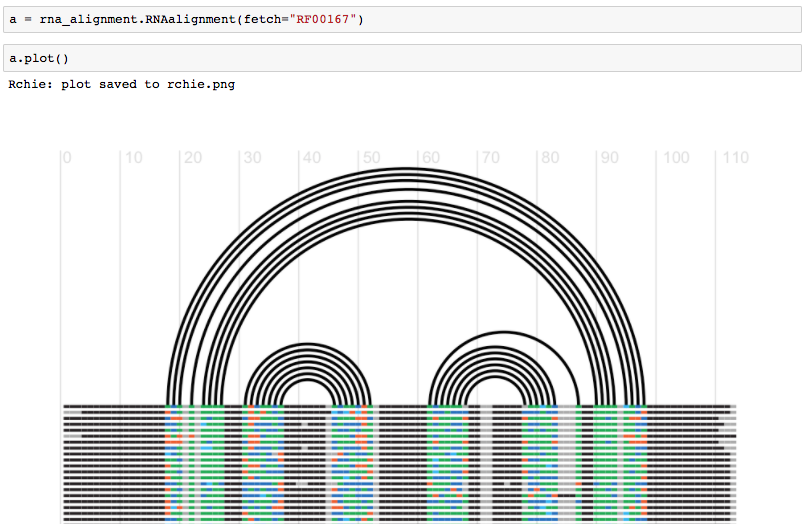
Fig. Fetch an alignment and generate an RChie plot for it. See more https://github.com/mmagnus/rna-tools/blob/master/rna_tools/tools/rna_alignment/rna_alignment.ipynb
Take a tour http://mmagnus.github.io/rna-tools/#/ and/or read the doc rna-tools.rtfd.io/en/latest/.

Fig. rna_pdb_toolsx.py --get-rnapuzzle-ready *pdb --inplace
(see CHANGELOG for more detailed description)
22-05-17 🔥 a paper published on our server 🔥
rna-tools.online: a Swiss army knife for RNA 3D structure modeling workflow
Marcin Magnus
Nucleic Acids Research
https://doi.org/10.1093/nar/gkac372
22-03-31 rna-tools goes online, http://rna-tools.online
21-05-24 spotifier branched out into own repository to keep RT light https://github.com/mmagnus/yeast-spotifier
20-10/12 mqapRNA: py3 wrappers and include them in RT: RASP, Dfire, RNA3DCNN, QRNA, FARNA(Rosetta), AnalyzeGeometry, ClashScore, eSCORE (barnaba), 3dRNAscore, RNAkb (5pt)
20-08-20 A new structures of splicesome processed with rna-tools to be easily viewed with PyMOL (or as single chains) PyMOL4Spliceosome
20-06-18 A new tool, spotifier, to process yeast plate images into figures
20-03-21 PyMOL Preview Generator a new tool for generation of previews in Finders created :-)
19-11-08 The RNA-Puzzles toolkit paper has been accepted for publication in Nucleic Acid Research :-) Release v3
RNA-Puzzles toolkit: A computational resource of RNA 3D structure benchmark datasets, structure manipulation, and evaluation tools Magnus, Marcin; Antczak, Maciej; Zok, Tomasz; Wiedemann, Jakub ; Lukasiak, Piotr; Cao, Yang ; Bujnicki, Janusz; Westhof, Eric; Szachniuk, Marta; Miao, Zhichao https://academic.oup.com/nar/advance-article/doi/10.1093/nar/gkz1108/5651330
19-10-22 We made a searchable index of all the tools. There are around 100 functionalities implemented, enjoy it! Let us know if something is missing or unclear!
19-10-10 rna-tools finally works with Python 3, to get Python 2 version go to this branch However, not all tools can be used with Python 3, for example, ClaRNA is written in Python 2 and we can do nothing about it. So, for now, we suggest using Conda or something else that supports kind of hybrid Python2/Python3 environments. Read more on this here and here
19-10-01 The EvoClustRNA manuscript with the heavy use of rna-tools is accepted for publication!
M. Magnus, M., Kappel, K., Das, R., & Bujnicki, J. M. (2019). RNA 3D structure prediction guided by independent folding of homologous sequences, BMC Bioinformatics, https://github.com/mmagnus/EvoClustRNA https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-019-3120-y
19-06-15 rna-tools used for spliceosome! :-) is accepted for publication! See this folder for the description of the analysis!
Eysmont, K., Matylla-Kulinska, K., Jaskulska, A., Magnus, M., & Konarska, M. M. (2018). Rearrangements within the U6 snRNA core at the transition between the two catalytic steps of splicing. Molecular Cell https://github.com/mmagnus/rna-tools/tree/master/U6MolCell https://www.cell.com/molecular-cell/pdfExtended/S1097-2765(19)30390-9
See also CHANGELOG.
- Tour
- rna_pdb_toolsx.py
- Tools
- Docs
- Cite
- Used in papers
- RNA Puzzle Submission
- Inspiration (and alternatives)
- Install
- Index of Tools
- Index of Jupyter Notebooks
- References
Take a tour http://mmagnus.github.io/rna-tools/#/
usage: rna_pdb_toolsx.py [-h] [--version] [-r] [--no-progress-bar] [--renum-atoms] [--renum-nmr]
[--renum-residues-dirty] [--undo] [--delete-anisou] [--fix] [--to-mol2]
[--split-alt-locations] [-c] [--is-pdb] [--is-nmr] [--nmr-dir NMR_DIR]
[--un-nmr] [--orgmode] [--get-chain GET_CHAIN] [--fetch] [--fetch-ba]
[--fetch-chain] [--get-seq] [--color-seq] [--ignore-files IGNORE_FILES]
[--compact] [--hide-warnings] [--get-ss] [--rosetta2generic] [--no-hr]
[--renumber-residues] [--dont-rename-chains] [--dont-fix-missing-atoms]
[--inspect] [--collapsed-view] [--cv] [-v] [--mutate MUTATE] [--edit EDIT]
[--rename-chain RENAME_CHAIN] [--swap-chains SWAP_CHAINS]
[--set-chain SET_CHAIN] [--replace-chain REPLACE_CHAIN] [--delete DELETE]
[--extract EXTRACT] [--extract-chain EXTRACT_CHAIN] [--uniq UNIQ]
[--chain-first] [--oneline] [--replace-htm] [--fasta] [--cif2pdb] [--pdb2cif]
[--mdr] [--get-rnapuzzle-ready] [--rpr] [--keep-hetatm] [--inplace]
[--suffix SUFFIX] [--replace-hetatm] [--dont-report-missing-atoms]
[--backbone-only] [--no-backbone] [--bases-only]
file [file ...]
rna_pdb_toolsx - a swiss army knife to manipulation of RNA pdb structures
Usage::
$ rna_pdb_toolsx.py --delete A:46-56 --inplace *.pdb
$ rna_pdb_toolsx.py --get-seq *
# BujnickiLab_RNApuzzle14_n01bound
> A:1-61
# BujnickiLab_RNApuzzle14_n02bound
> A:1-61
CGUUAGCCCAGGAAACUGGGCGGAAGUAAGGCCCAUUGCACUCCGGGCCUGAAGCAACGCG
[...]
-v is for verbose, --version for version ;)
positional arguments:
file file
optional arguments:
-h, --help show this help message and exit
--version
-r, --report get report
--no-progress-bar for --no-progress-bar for --rpr
--renum-atoms renumber atoms, tested with --get-seq
--renum-nmr
--renum-residues-dirty
--undo undo operation of action done --inplace, , rename "backup files" .pdb~ to pdb, ALL files in the folder, not only ~ related to the last action (that you might want to revert, so be careful)
--delete-anisou remove files with ANISOU records, works with --inplace
--fix fix a PDB file, ! external program, pdbfixer used to fix missing atoms
--to-mol2 fix a PDB file, ! external program, pdbfixer used to fix missing atoms
--split-alt-locations
@todo
-c, --clean get clean structure
--is-pdb check if a file is in the pdb format
--is-nmr check if a file is NMR-style multiple model pdb
--nmr-dir NMR_DIR make NMR-style multiple model pdb file from a set of files
rna_pdb_toolsx.py --nmr-dir . 'cwc15_u5_fragments*.pdb' > ~/Desktop/cwc15-u5.pdb
please use '' for pattern file recognition, this is a hack to deal with folders with
thousands of models, if you used only *.pdb then the terminal will complain that you
selected to many files.
--un-nmr split NMR-style multiple model pdb files into individual models [biopython]
--orgmode get a structure in org-mode format <sick!>
--get-chain GET_CHAIN
get chain, one or many, e.g, A, but now also ABC works
--fetch fetch file from the PDB db, e.g., 1xjr,
use 'rp' to fetchthe RNA-Puzzles standardized_dataset [around 100 MB]
--fetch-ba fetch biological assembly from the PDB db
--fetch-chain fetch a structure in extract chain, e.g. 6bk8 H
--get-seq get seq
--color-seq color seq, works with --get-seq
--ignore-files IGNORE_FILES
files to be ingored, .e.g, 'solution'
--compact with --get-seq, get it in compact view'
$ rna_pdb_toolsx.py --get-seq --compact *.pdb
# 20_Bujnicki_1
ACCCGCAAGGCCGACGGCGCCGCCGCUGGUGCAAGUCCAGCCACGCUUCGGCGUGGGCGCUCAUGGGU # A:1-68
# 20_Bujnicki_2
ACCCGCAAGGCCGACGGCGCCGCCGCUGGUGCAAGUCCAGCCACGCUUCGGCGUGGGCGCUCAUGGGU # A:1-68
# 20_Bujnicki_3
ACCCGCAAGGCCGACGGCGCCGCCGCUGGUGCAAGUCCAGCCACGCUUCGGCGUGGGCGCUCAUGGGU # A:1-68
# 20_Bujnicki_4
--hide-warnings hide warnings, works with --get-chain, it hides warnings that given changes are not detected in a PDB file
--get-ss get secondary structure
--rosetta2generic convert ROSETTA-like format to a generic pdb
--no-hr do not insert the header into files
--renumber-residues by defult is false
--dont-rename-chains used only with --get-rnapuzzle-ready.
By default:
--get-rnapuzzle-ready rename chains from ABC.. to stop behavior switch on this option
--dont-fix-missing-atoms
used only with --get-rnapuzzle-ready
--inspect inspect missing atoms (technically decorator to --get-rnapuzzle-ready without actually doing anything but giving a report on problems)
--collapsed-view
--cv alias to collapsed_view
-v, --verbose tell me more what you're doing, please!
--mutate MUTATE mutate residues,
e.g.,
--mutate "A:1a+2a+3a+4a,B:1a"
to mutate to adenines the first four nucleotides of the chain A
and the first nucleotide of the chain B
--edit EDIT edit 'A:6>B:200', 'A:2-7>B:2-7'
--rename-chain RENAME_CHAIN
edit 'A>B' to rename chain A to chain B
--swap-chains SWAP_CHAINS
B>A, rename A to _, then B to A, then _ to B
--set-chain SET_CHAIN
set chain for all ATOM lines and TER (quite brutal function)
--replace-chain REPLACE_CHAIN
a file PDB name with one chain that will be used to
replace the chain in the original PDB file,
the chain id in this file has to be the same with the chain id of the original chain
--delete DELETE delete the selected fragment, e.g. A:10-16, or for more than one fragment --delete 'A:1-25+30-57'
--extract EXTRACT extract the selected fragment, e.g. A:10-16, or for more than one fragment --extract 'A:1-25+30-57'
--extract-chain EXTRACT_CHAIN
extract chain, e.g. A
--uniq UNIQ
rna_pdb_toolsx.py --get-seq --uniq '[:5]' --compact --chain-first * | sort
A:1-121 ACCUUGCGCAACUGGCGAAUCCUGGGGCUGCCGCCGGCAGUACCC...CA # rp13nc3295_min.out.1
A:1-123 ACCUUGCGCGACUGGCGAAUCCUGAAGCUGCUUUGAGCGGCUUCG...AG # rp13cp0016_min.out.1
A:1-123 ACCUUGCGCGACUGGCGAAUCCUGAAGCUGCUUUGAGCGGCUUCG...AG # zcp_6537608a_ALL-000001_AA
A:1-45 57-71 GGGUCGUGACUGGCGAACAGGUGGGAAACCACCGGGGAGCGACCCGCCGCCCGCCUGGGC # solution
--chain-first
--oneline
--replace-htm
--fasta with --get-seq, show sequences in fasta format,
can be combined with --compact (mind, chains will be separated with ' ' in one line)
$ rna_pdb_toolsx.py --get-seq --fasta --compact input/20_Bujnicki_1.pdb
> 20_Bujnicki_1
ACCCGCAAGGCCGACGGC GCCGCCGCUGGUGCAAGUCCAGCCACGCUUCGGCGUGGGCGCUCAUGGGU
--cif2pdb [PyMOL Python package required]
--pdb2cif [PyMOL Python package required]
--mdr get structures ready for MD (like rpr but without first)
RNAPUZZLE-READY:
--get-rnapuzzle-ready
get RNApuzzle ready (keep only standard atoms).'
Be default it does not renumber residues, use --renumber-residues
[requires BioPython]
--rpr alias to get_rnapuzzle ready)
CAN BE COMBINED WITH:
--keep-hetatm keep hetatoms
--inplace in place edit the file! [experimental,
only for get_rnapuzzle_ready, --delete, --get-ss, --get-seq, --edit-pdb]
--suffix SUFFIX when used with --inplace allows you to change a name of a new file, --suffix del will give <file>_del.pdb (mind added _)
--replace-hetatm replace 'HETATM' with 'ATOM' [tested only with --get-rnapuzzle-ready]
--dont-report-missing-atoms
used only with --get-rnapuzzle-ready
--backbone-only used only with --get-rnapuzzle-ready, keep only backbone (= remove bases)
--no-backbone used only with --get-rnapuzzle-ready, remove atoms of backbone (define as P OP1 OP2 O5')
--bases-only used only with --get-rnapuzzle-ready, keep only atoms of bases
Tricks:
$ rna_pdb_toolsx.py --delete A:48-52 --suffix=noloop --inplace
10_rp17c.out.14.pdb
10_rp17c.out.14_out.pdb
[..]
$ rna_pdb_toolsx.py --get-rnapuzzle-ready --inplace *.pdb
.. keep original structures in original and use rpr:
➜ bujnicki_server_ss for i in original/*.pdb; do rna_pdb_toolsx.py --get-rnapuzzle-ready $i > ${i/.pdb/_rpr.pdb}; done
➜ bujnicki_server_ss ls
17pz_withSS_all_thrs6.00A_clust01-000001_AA_rpr.pdb 17pz_withSS_all_thrs6.00A_clust06-000001_AA_rpr.pdb
17pz_withSS_all_thrs6.00A_clust02-000001_AA_rpr.pdb 17pz_withSS_all_thrs6.00A_clust07-000001_AA_rpr.pdb
17pz_withSS_all_thrs6.00A_clust03-000001_AA_rpr.pdb 17pz_withSS_all_thrs6.00A_clust08-000001_AA_rpr.pdb
17pz_withSS_all_thrs6.00A_clust04-000001_AA_rpr.pdb 17pz_withSS_all_thrs6.00A_clust09-000001_AA_rpr.pdb
17pz_withSS_all_thrs6.00A_clust05-000001_AA_rpr.pdb original
.. or to get structures ready for SimRNA/SimRNAweb use :
$ for i in *pdb; do rna_pdb_toolsx.py --get-rnapuzzle-ready $i > ${i/.pdb/_srr.pdb}; done
# at some point there was a seperate function --get_simrna_ready but there is no need for it
# simply use --get-rnapuzzle-ready
rna-tools loves to be used with parallel (wirte in shell script what you want rna-tools to do) and run to execute it in a set of folders:
parallel --bar --eta --progress 'cp test.sh {} && cd {} && bash test.sh ' ::: *
The (almost) full list of tools can be found here: https://github.com/mmagnus/rna-tools/blob/master/rna-tools-index.csv
Read more http://rna-tools.readthedocs.io/en/latest/
Read the documentations at rna-tools.rtfd.io/en/latest/.

Magnus M, Antczak M, Zok T, Wiedemann J, Lukasiak P, Cao Y, Bujnicki JM, Westhof E, Szachniuk M, Miao Z. RNA-Puzzles toolkit: a computational resource of RNA 3D structure benchmark datasets, structure manipulation, and evaluation tools.
Nucleic Acids Research. 2019
10.1093/nar/gkz1108
https://academic.oup.com/nar/advance-article/doi/10.1093/nar/gkz1108/5651330
The papers in which the rna-tools package was used or one of spin-off projects, e.g., RNA-Puzzles-Normalized-submissions, PyMOL4Spliceosome:
[12] C. van der Feltz, B. Nikolai, C. Schneider, J. C. Paulson, X. Fu, and A. A. Hoskins, “Saccharomyces cerevisiaeEcm2 Modulates the Catalytic Steps of pre-mRNA Splicing,” bioRxiv, vol. 4, pp. 2132–45, Sep. 2020.
[11] T. Zhang, G. Hu, Y. Yang, J. Wang, and Y. Zhou, “All-Atom Knowledge-Based Potential for RNA Structure Discrimination Based on the Distance-Scaled Finite Ideal-Gas Reference State.,” J. Comput. Biol., vol. 27, no. 6, pp. 856–867, Jun. 2020.
[10] F. Stefaniak and J. M. Bujnicki, “AnnapuRNA: a scoring function for predicting RNA-small molecule interactions.,” biorxiv.org 2020 https://github.com/filipsPL/annapurna
[9] G. Chojnowski, M. Magnus, and J. M. Bujnicki, “RNA fragment assembly with experimental restraints,” (in progress) Jun. 2020. http://iimcb.genesilico.pl/rnamasonry
[8] A. M. Watkins, R. Rangan, and R. Das, “FARFAR2: Improved De Novo Rosetta Prediction of Complex Global RNA Folds.,” Structure, Jun. 2020.
[7] Z. Miao, R. W. Adamiak, M. Antczak, M. J. Boniecki, J. M. Bujnicki, S.-J. Chen, C. Y. Cheng, Y. Cheng, F.-C. Chou, R. Das, N. V. Dokholyan, F. Ding, C. Geniesse, Y. Jiang, A. Joshi, A. Krokhotin, M. Magnus, O. Mailhot, F. Major, T. H. Mann, P. Piatkowski, R. Pluta, M. Popenda, J. Sarzynska, L. Sun, M. Szachniuk, S. Tian, J. Wang, J. Wang, A. M. Watkins, J. Wiedemann, Y. Xiao, X. Xu, J. D. Yesselman, D. Zhang, Y. Zhang, Z. Zhang, C. Zhao, P. Zhao, Y. Zhou, T. Zok, A. Zyła, A. Ren, R. T. Batey, B. L. Golden, L. Huang, D. M. Lilley, Y. Liu, D. J. Patel, and E. Westhof, “RNA-Puzzles Round IV: 3D structure predictions of four ribozymes and two aptamers.,” RNA, p. rna.075341.120, May 2020.
[6] M. Magnus, K. Kappel, R. Das, and J. M. Bujnicki, “RNA 3D structure prediction guided by independent folding of homologous sequences.,” BMC Bioinformatics, vol. 20, no. 1, pp. 512–15, Oct. 2019. https://github.com/mmagnus/EvoClustRNA
[5] K. Eysmont, K. Matylla-Kulinska, A. Jaskulska, M. Magnus, and M. M. Konarska, “Rearrangements within the U6 snRNA Core during the Transition between the Two Catalytic Steps of Splicing.,” Molecular Cell, vol. 75, no. 3, pp. 538–548.e3, Aug. 2019. https://github.com/mmagnus/rna-tools/tree/master/U6MolCell
[4] J. Li, W. Zhu, J. Wang, W. Li, S. Gong, J. Zhang, and W. Wang, “RNA3DCNN: Local and global quality assessments of RNA 3D structures using 3D deep convolutional neural networks.,” PLoS Comput Biol, vol. 14, no. 11, p. e1006514, Nov. 2018. http://doi.org/10.1371/journal.pcbi.1006514
[3] P. Boccaletto, M. Magnus, C. Almeida, A. Zyła, A. Astha, R. Pluta, B. Bagiński, E. J. Jankowska, S. Dunin-Horkawicz, T. K. Wirecki, M. J. Boniecki, F. Stefaniak, and J. M. Bujnicki, “RNArchitecture: a database and a classification system of RNA families, with a focus on structural information.,” Nucleic Acids Research, vol. 46, no. 1, pp. D202–D205, Jan. 2018. https://iimcb.genesilico.pl/RNArchitecture/
[2] M. Magnus, M. J. Boniecki, W. K. Dawson, and J. M. Bujnicki, “SimRNAweb: a web server for RNA 3D structure modeling with optional restraints.,” Nucleic Acids Research, vol. 44, no. 1, pp. W315–9, Jul. 2016. https://iimcb.genesilico.pl/SimRNAweb/
[1] I. Tuszyńska, M. Magnus, K. Jonak, W. K. Dawson, and J. M. Bujnicki, “NPDock: a web server for protein-nucleic acid docking.,” Nucleic Acids Research, vol. 43, no. 1, pp. W425–30, Jul. 2015. http://iimcb.genesilico.pl/NPDock/
Read at https://rna-tools.readthedocs.io/en/latest/rna-puzzles.html
- https://www.rosettacommons.org/docs/latest/application_documentation/rna/RNA-tools
- http://blue11.bch.msu.edu/mmtsb/convpdb.pl
- https://github.com/haddocking/pdb-tools
- https://github.com/harmslab/pdbtools
- http://ginsberg.med.virginia.edu/Links/Phenix/pdbtools.htm
- an amazing PDB fixer! https://github.com/openmm/pdbfixer
- .. and more!
pip install rna-tools
For developers see this http://rna-tools.readthedocs.io/en/latest/install-dev.html
The index in a form of a searchable table can be found here.
--get-rnapuzzle-readyformat PDB file to be compatible with the "RNA-Puzzle PDB format",--reportget report--renum-atomsrenumber atoms, tested with --get-seq--renum-residues-dirty--renumber-residuesby defult is false--delete-anisouremove files with ANISOU records, works with --inplace--split-alt-locations--cleanget clean structure--is-pdbcheck if a file is in the pdb format--is-nmrcheck if a file is NMR-style multiple model pdb--un-nmrSplit NMR-style multiple model pdb files into individual models [biopython]--orgmodeget a structure in org-mode format <sick!>--get-chain GET_CHAIN--fetchfetch file from the PDB db--fetch-bafetch biological assembly from the PDB db--get-seqget seq--compactwith --get-seq, get it in compact view'--get-ssget secondary structure--rosetta2genericconvert ROSETTA-like format to a generic pdb--get-rnapuzzle-ready--collapsed-view--replace-hetatmreplace 'HETATM' with 'ATOM' [tested only with --get-rnapuzzle-ready]--mutate MUTATEmutate residues,--edit EDITedit 'A:6>B:200', 'A:2-7>B:2-7'--rename-chain RENAME_CHAIN--swap-chains SWAP_CHAINS--replace-chain REPLACE_CHAIN--delete DELETEdelete the selected fragment, e.g. A:10-16, or for more than one fragment --delete 'A:1-25+30-57'--extract EXTRACTextract the selected fragment, e.g. A:10-16, or for more than one fragment --extract 'A:1-25+30-57'--extract-chain EXTRACT_CHAIN
BlastPDB.py- a simple Blast search,RfamSearch.py- a simple Rfam search.
rna_secondary_structure_prediction.py- a wrapper for secondary structure prediction methods, e.g., cyclefold, mcfold,ipknot, RNAsubopt, contextfold, centroid_fold, with a use of restraints (if applicable)rna_dot2ct.py- convert dot notation to ct notation.- secondary structure format conversion tools
rna_calc_rmsd.py- calculate RMSDs of structures to the targetrna_calc_evo_rmsd.py- calculate RMSD between structures based on a given alignment and selected residues as defined in the "x line",rna_calc_inf.py- including multiprocessing based on ClaRNA (in Python 2!)rna_clanstix.py- a tool for visualizing RNA 3D structures based on pairwise structural similarity with Clans,rna_prediction_significance.py- calculate significance of an RNA tertiary structure prediction.
-
<a href="https://rna-tools.readthedocs.io/en/latest/tools.html#module-rna_tools.tools.diffpdb.diffpdb>
diffpdb- a simple tool to compare text-content of PDB files, -
rna_pdb_merge_into_one.py- merge single files into an NMR-style multiple model file PDB file.
clarna_app.py- a wrapper to ClaRNA, See also PyMOL4RNA, Python 2!rna_x3dna.py- a wrapper to 3dna, See also PyMOL4RNA,ClashCalc.py- a simple clash score calculator, used in NPDock, requires BioPython,
rna_refinement.py- a wrapper for QRNAS (Quick Refinement of Nucleic Acids)
- Undo ("Quick Save & Load") for PyMOL,
CTRL-S&CTRL-Z, - PyMOL4Spliceosome (link to its own repository)
clarna()- contact classification with ClaRNA directly in PyMOL for selected residues,x3dna()- contact classification with X3DNA directly in PyMOL for selected residues,ss()- get secondary structures of selected objects,sav <fn>- save on Desktop a session and a PNG file illustrating the session,- color structure domains according to pre-defined styles, e.g.,
rp17() - PyMOL Preview Generator for OSX
rna_simrna_cluster.pyrna_simrna_extract.pyrna_simrna_get_datarna_simrna_lowest.py- SimRNAweb:
rna_simrnaweb_download_job.py- download model files, trajectory for a given SimRNAweb job rna_pdb_merge_structure_with_fragments.py- insert fragments into the structure, used at the SimRNAweb server for modeling with a given pre-define structure,rna_pdb_edit_occupancy_bfactor.py- edit occupancy or bfactor in PDB file,rna_pk_simrna_to_one_line.py- convert multi-line SimRNA secondary structure format to one line bracket format,rna_ss_pk_to_simrna.py- do opposite as previous one, convert one line bracket format with pseudoknots into multi-line SimRNA secondary structure format,- See also
simrna_trajectoryin Python Classes.
rna_rosetta_n.pyrna_rosetta_check_progress.pyrna_rosetta_min.pyrna_rosetta_cluster.pyrna_rosetta_extract_lowscore_decoys.pyrna_rosetta_run.pyrna_rosetta_head.py
get_seq()- get sequence,get_ss()- get secondary structure for a given sequence,fetch()- fetch an alignment from Rfam,cmalign()- aligns the RNA sequences in to the covariance model (CM) inRchie()- plotting arc diagrams of RNA secondary structures,find_core()- finds core of molecules in alignment,
Seq.py- seq processing, including secondary structure predictionSecondaryStructure.py::draw_ss()SecondaryStructure.py::parse_vienna_to_pairs()simrna_trajectory
- rnakb_utils - RNAkb-related tools,
- rnapuzzle_sender - a script to send PDB files to the RNA-Puzzle organizers,
- rnashape2ascii - convert RNA shape data into ascii characters ;-)
▅▄▆▄▂▁▁▁▁▁▁▁▁▁▁▂▁▁▁▁▁▁▁▁▁▁▁▁▁▁▁▁▂▅▇▅▄▃▂▁ - cluster_load - scripts to view cluster load, based on processing
qstat.
- https://github.com/mmagnus/rna-tools/blob/master/notes/fig3-manuscript.ipynb
- https://github.com/mmagnus/rna-tools/blob/master/rna_tools/tools/rna_alignment/rna_alignment.ipynb
Components of rna-tools are based upon the following pieces of scientific literature:
P. J. A. Cock, T. Antao, J. T. Chang, B. A. Chapman, C. J. Cox, A. Dalke, I. Friedberg, T. Hamelryck, F. Kauff, B. Wilczynski, and M. J. L. de Hoon, “Biopython: freely available Python tools for computational molecular biology and bioinformatics.,” Bioinformatics, vol. 25, no. 11, pp. 1422–1423, Jun. 2009.
M. Rother, K. M. Rother, T. Puton, and J. M. Bujnicki, “ModeRNA: a tool for comparative modeling of RNA 3D structure.,” Nucleic Acids Research, vol. 39, no. 10, pp. 4007–4022, May 2011.
T. Waleń, G. Chojnowski, P. Gierski, and J. M. Bujnicki, “ClaRNA: a classifier of contacts in RNA 3D structures ased on a comparative analysis of various classification schemes.,” Nucleic Acids Research, vol. 42, no. 19, pp. e151–e151, Oct. 2014.
and more, see seperate readmes.